
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 56, Num. 3, 2008, pp. 236-247
|
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 236-247
Review Article
Duchenne muscular dystrophy
Yiu EppieM, Kornberg AndrewJ
Children's Neuroscience Centre, Royal Children's Hospital Melbourne; Murdoch Children's Research Institute; Department of Pediatrics University of Melbourne
Correspondence Address:Children's Neuroscience Centre, Royal Children's Hospital Melbourne, Flemington Road, Parkville, Victoria, 3052
andrew.kornberg@rch.org.au
Date of Acceptance: 26-Sep-2008
Code Number: ni08073
Abstract Duchenne muscular dystrophy (DMD), an X-linked disorder, is the most common muscular dystrophy in children, presenting in early childhood and characterized by proximal muscle weakness and calf hypertrophy in affected boys. Patients usually become wheelchair-bound by the age of 12 years, and die of cardiorespiratory complications in their late teens to early twenties. Advances in the management of DMD, including treatment with corticosteroids and the use of intermittent positive pressure ventilation have provided improvements in function, ambulation, quality of life and life expectancy, although novel therapies still aim to provide a cure for this devastating disorder. The clinical features, investigations, and management of DMD are reviewed, as well as the latest in some of the novel therapies.
Keywords: Continuous positive airway pressure, corticosteroids, creatine kinase, Duchenne muscular dystrophy, gene therapy, muscle disease, pediatric
Introduction Duchenne muscular dystrophy (DMD), an X-linked disorder, is the most common muscular dystrophy in children, presenting in early childhood and characterized by proximal muscle weakness and calf hypertrophy in affected boys. Patients usually become wheelchair-bound by 12 years and die in their late teens to early twenties. Advances in the management of this disorder with supportive therapy, corticosteroids, as well as novel therapies hope to change the outcomes of this disorder.
Clinical Features
Presenting features
The incidence of DMD is approximately 1 in 3500 live male births. [1]
The most frequent presenting symptoms are motor delay or an abnormal gait. Affected boys may present with difficulty in running or getting up from the ground, frequent falls, or toe-walking. Most present between three to five years. Less frequent presentations include language or global developmental delay, or incidentally raised serum creatine kinase or transaminase levels when these investigations are performed for other reasons.
Weakness
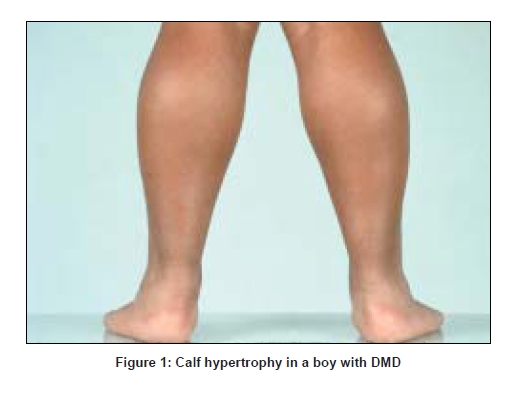
Proximal weakness affects the lower before the upper extremities, with progression to the point of wheelchair dependence. Eventually distal lower and then upper limb weakness occurs. Weakness of neck flexors is often present at presentation, and most patients with DMD have never been able to jump. Patients have a waddling gait, calf enlargement, and lumbar lordosis which disappears on sitting. Most become wheelchair-bound by age 11-12 years [2]. [Figure 1]
Cardiomyopathy
Cardiac disease consists of dilated cardiomyopathy due to cardiac fibrosis as well as disturbances of rhythm and conduction. [3],[4],[5]
Clinically apparent cardiomyopathy is first evident after 10 years of age, affects one-third of patients by age 14 years, and is present in all patients over 18 years of age. [5] Preclinical cardiac involvement is seen in 25% of patients under six years of age, with a persistent tachycardia commonly noted. [5] Atrial and ventricular arrhythmias occur, including premature ventricular beats and more complex or sustained ventricular ectopy, which increase with age and ventricular dysfunction. [4] Despite the high frequency of cardiac involvement, most patients are relatively asymptomatic due to physical inactivity. [5]
Respiratory complications
Chronic respiratory insufficiency due to restrictive lung disease is inevitable in all patients. Vital capacity increases as predicted until around age 10 years; after this time it starts to decrease at a rate of 8-12% per year. [6],[7],[8],[9] When vital capacity reaches less than 1 liter the risk of death within the next one to two years is relatively high. [6]
Obstructive sleep apnea is the predominant cause of sleep disordered breathing in the first decade, occurring in up to one-third of patients, with hypoventilation occurring in the second decade. [10] Four stages of hypercapnic chronic respiratory failure are typically described: Stage 1, sleep disordered breathing without hypercapnia; Stage 2, sleep disordered breathing with hypercapnia during rapid eye movement (REM) sleep; Stage 3, with hypercapnia during REM and Non-REM sleep; and Stage 4, diurnal hypercapnia. [10],[11],[12],[13],[14] At Stage 4, mean survival is less than 12 months without respiratory support. [15]
Intellectual disability
Intellectual disability is seen in 30% of boys with DMD, with the average intelligence quotient (IQ) being 85, normally distributed one standard deviation below the population norms. [16],[17],[18] Verbal IQ is more impaired than performance IQ. [16,19] Intellectual disability is not correlated with the severity of weakness. [16],[20],[21] Boys with DMD also have a higher incidence of attention deficit hyperactivity disorder [22],[23]
Orthopedic complications
Scoliosis develops in almost all children with DMD, and impacts on respiratory vital capacity. [24] It progresses significantly after boys lose ambulation, and maintenance of ambulation slows the rate of progression. [25] Long bone fractures are common and usually due to falls, affecting 21-44% of boys. [26],[27] Half of the fractures occur in independently ambulatory boys, with 20-40% losing ambulation as a result. [26],[27] Osteoporosis is present in most children with DMD. [27],[28],[29] Loss of bone mineral density begins even when boys are still ambulant, [27],[30] and continues to diminish with age.
Malignant hyperthermia
Whilst the association of malignant hyperthermia with DMD is not clearly established, patients with DMD are thought to have increased risk of malignant hyperthermia, or at least malignant hyperthermia-like reactions if exposed to inhalational anesthetics such as halothane, or succinylcholine. [31],[32],[33],[34]
Pathogenesis and Genetics
DMD is caused by mutations in the DMD gene, [35],[36] one of the largest known genes in humans, spanning 2.3 megabases and accounting for 0.1% of the total human genome. [37],[38] This gene encodes a protein called dystrophin, [39] which localizes to the cytoplasmic face of the sarcolemma (plasma membrane) of the skeletal muscle, [40] forming one component of a large glycoprotein complex (dystrophin-associated glycoprotein complex). [41],[42] Dystrophin consists of an N-terminal actin-binding domain, 24 spectrin-like repeat units interspersed by four hinge regions, followed by a cysteine-rich domain and a C-terminal domain. [39],[43] The cysteine-rich domain binds to laminin-2 via alpha and beta dystroglycan, and therefore acts as mechanical link between actin in the cytoskeleton, and the extracellular matrix. [42],[44],[45]
The DMD gene contains 79 exons, but accounts for only 0.6% of the gene; the rest made of large introns.[46] The large size of the DMD gene makes it susceptible to mutations, with one-third of all mutations arising de novo .
Mutations in the DMD gene result in loss of function of dystrophin, resulting in a prematurely truncated, unstable dystrophin protein. The reading-frame rule explains the phenotypic differences between DMD and Becker muscular dystrophy (BMD): mutations that disrupt the open reading frame, resulting in an abnormal and truncated dystrophin cause DMD, whilst mutations which maintain the open reading frame, resulting in a shorter lower molecular weight, but partly functional dystrophin, cause BMD. [47],[48] Ninety per cent of cases of DMD and BMD conform to this reading-frame rule. [48]
The majority of mutations are intragenic deletions, which account for 65-72% of all DMD patients. [48],[49] Most deletions occur in the ′hotspot′ region, spanning Exons 45-53, [50],[51] with the most common deletions being of Exon 45 and Exons 45-47. [48] Single or multiexon duplications are found in 7% of patients, [48],[52] most located in a minor hotspot spanning Exons 2-20. [50],[51] Point mutations, small deletions or insertions account for 20% of patients without deletions or duplications. Most are nonsense, frameshift or splice site mutations; missense mutations are extremely rare. [48],[53]
The precise mechanism of how dystrophin deficiency leads to degeneration of muscle fibers remains unclear. Absence of dystrophin at the plasma membrane leads to delocalisation of dystrophin-associated proteins from the membrane, disruption of the cytoskeleton with resultant membrane instability and increased susceptibility to mechanical stress. [41],[54],[55] In addition, altered membrane permeability and abnormal calcium homeostasis are thought to play a role, with increased cytosolic calcium concentration leading to activation of proteases such as calpains. [55],[56],[57],[58] The absence of nitric oxide synthase, delocalized from the subsarcolemmal membrane, may contribute to damage, but is not thought to directly cause dystrophic features. [59],[60],[61]
Various animal models of DMD exist; the most commonly studied and used is the mdx mouse. Several strains of mdx mouse have been characterized; the most commonly used strain has a nonsense mutation in Exon 23 of the DMD gene. [62],[63]
Investigations
Serum muscle enzymes
The characteristic finding in DMD is a markedly raised serum CK level, at least 10 to 20 times (and often 50 to 200 times) the upper limit of normal before the age of five years. Serum CK concentrations are high even in newborns and prior to any symptoms. The high CK levels at birth can form the basis of neonatal screening for DMD. Levels peak at two to three years of age and then decline with increasing age, due to progressive loss of dystrophic muscle fibers. [64],[65],[66] A serum CK less than 10 times normal in a child with suspected DMD in the first three years of life should raise the question of an alternate diagnosis. [2]
Serum alanine transaminase and aspartate transaminase levels are raised in DMD and tend to correlate with CK levels. [67],[68],[69] Other enzymes raised in DMD include aldolase and lactate dehydrogenase. [70],[71],[72] Most of these are not specific for muscle and are generally not useful in the diagnosis of DMD.
Electromyography
Electromyography and nerve conduction studies are rarely required in the diagnosis of DMD. Needle electromyography findings are myopathic, with short duration, low amplitude polyphasic motor unit potentials, particularly in proximal muscles. [2] Abnormal spontaneous activity in the form of fibrillation potentials, positive sharp waves and complex repetitive discharges may be detected due to denervation and some reinnervation in necrotic muscles. This may also result in the presence of satellite motor unit potentials.[73],[74] Over time the motor units become very small and some areas become electrically silent. [2]
Nerve conduction studies are normal in early DMD. As the disease progresses, compound muscle action potentials decrease in amplitude.
Muscle biopsy
In some centers, muscle biopsy is no longer routine in the diagnostic workup of DMD if genetic testing is positive and the clinical phenotype is consistent. Muscle biopsy is then only performed where genetic testing is negative, or the clinical phenotype is atypical. Others, however, advocate for muscle biopsy to remain a routine investigation in DMD, as it remains the gold standard for diagnosis. [75]
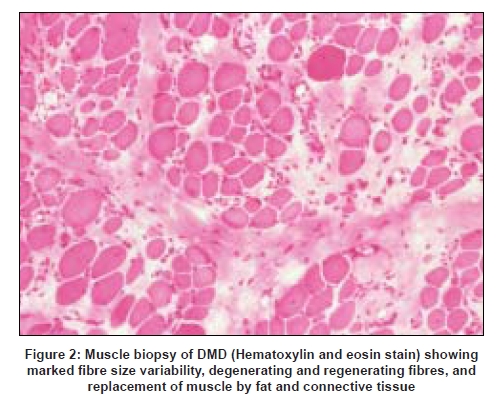
On light microscopy early changes include degenerating necrotic muscle fibers with invasion by macrophages, as well as clusters of small- to intermediate-sized regenerating muscle fibers which have basophilic cytoplasm. [2] Increased variability of muscle fiber size is also seen, initially with larger than normal, then smaller than normal fibers as the disease progresses. [76] Type 1 fiber predominance is observed, as are hypercontracted muscle fibers. Eventually there is significant replacement of muscle fibers by fat and endomysial connective tissue [Figure 2].
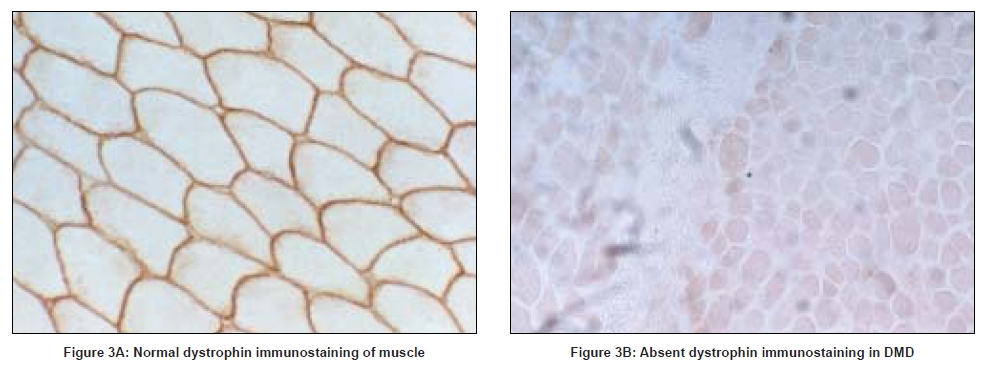
Absent or markedly reduced dystrophin in muscle biopsies of boys with DMD can be demonstrated on immunostaining and/or Western blot analysis, using antibodies directed against different epitopes of dystrophin. Generally, antibodies recognizing the amino-terminus, carboxy-terminus and rod domains are used. Immunostaining using the amino-terminus or rod domain antibodies shows faint sarcolemmal staining in up to 60% of DMD patients. Immunoreactivity to carboxy-terminal antibodies however is absent in DMD and is therefore useful in differentiating DMD from BMD. [77],[78],[79] Western blot analysis allows quantification of the amount of dystrophin protein as well assessment of size of the protein present. In DMD less than 5% of the normal quantity of dystrophin is present when carboxy-terminal antibodies are used, whilst up to 25% of normal dystrophin levels is seen with the use of rod domain antibodies. [2],[80],[81] [Figure 3].
Molecular genetic testing
Molecular genetic testing is now the mainstay of diagnosis in most centers. A multiplex polymerase chain reaction (PCR), covering 18 exons at the deletion hotspots developed by Chamberlain and Beggs detected 90-98% of all deletions, although duplications were not identified by this method. [50],[82] More recently, the development of multiplex ligation-dependent probe amplification (MLPA) has provided a more sensitive technique for detecting deletions. All 79 exons are covered by two sets of probes, with individual exons depicted as a single peak. This allows gene dosage abnormalities to be detected, allowing detection of duplications and testing of carrier individuals as well as for deletions. [83],[84],[85],[86] Occasionally, point mutations will also be detected as single exon deletions, with further analysis allowing more specific delineation of the point mutation. [83]
If MLPA testing is negative, the DMD gene can be tested for point mutations. Direct sequence analysis of the DMD gene is generally available on a research basis only, due to its labor-intensive and costly nature. Several groups have developed strategies to target exonic regions for direct sequencing after the use of initial screening methods. [87],[88]
A targeted high-density oligonucleotide comparative genomic hybridization microarray that allows high-resolution analysis of the DMD gene has also been developed recently, allowing identification of deletions and duplications but also previously unidentified deep intronic mutations. [89]
Muscle magnetic resonance imaging
Muscle MRI is usually not performed in DMD for diagnosis, but may be a useful noninvasive tool to evaluate progression of muscle involvement over time. Abnormalities in signal are seen on T1 and T2 images, with initial selective involvement of the gluteus maximus, adductor magnus, quadriceps, biceps femoris, rectus femoris and gastrocnemii muscles. [90]
Carrier Females
The majority of female DMD carriers are asymptomatic. However, 2-20% of carriers have clinically evident muscle weakness. [91],[92] Weakness is usually mild to moderate, and asymmetric. [91] Some carriers report myalgia or cramps without weakness. [91] Creatine kinase levels are raised in 50-60% of known carriers. [91] Cardiac involvement in carrier females is well recognized, but is usually subclinical, although severe heart failure has been reported. [93],[94] Dilated cardiomyopathy was present in 8% of carriers in one cross-sectional study, with another 19% of carriers with left ventricular dilatation. [93] Despite this, however, these carriers do not appear to have reduced life expectancy or increased risk of cardiac death. [95] The age of onset of cardiomyopathy is unclear, but is thought to be in the early adult years.[96] There is no association between the degree of muscle weakness and degree of cardiomyopathy. [91]
Around 20% of carriers have abnormal dystrophin immunostaining on muscle biopsy, with a mosaic pattern of dystrophin-positive and dystrophin-negative fibers present. [97],[98] No association has been found between dystrophin abnormalities and the presence or absence of muscle weakness or cardiomyopathy. [98]
Management
General management and surveillance
Symptomatic management and surveillance of the orthopedic, cardiac and respiratory complications associated with DMD allows anticipation, early detection and treatment of these complications.
Cardiac disease
Cardiac surveillance should commence at approximately 10 years of age and continue on an annual or biannual basis. Evaluation should include an electrocardiogram and transthoracic echocardiogram, with consideration of cardiac magnetic resonance imaging in patients with limited echocardiographic acoustic windows. Periodic Holter monitoring should also be considered in patients with known cardiac rhythm abnormalities. [99]
There is a trend for early treatment of dilated cardiomyopathy with angiotensin converting enzyme inhibitors and beta blockers in DMD, with normalization or improvement in left ventricular dysfunction demonstrated in some patients, thought to be due to cardiac remodeling. [100],[101] Prospective trials are required to evaluate further the optimal management of cardiomyopathy in DMD.
Respiratory disease
Baseline pulmonary function tests and respiratory evaluations should begin at age 8 to 9 years and before ambulation is lost. Ongoing evaluations should occur annually and then biannually after wheelchair confinement. In addition to spirometry, early morning and daytime carbon dioxide levels should be measured. If available, annual polysomnography to detect sleep disordered breathing and nocturnal hypoventilation should also be performed on an annual basis from the time of loss of ambulation. All patients should receive the pneumococcal vaccine and an annual influenza vaccination. Effective airway clearance methods, including the use of manual and mechanical techniques (e.g. mechanical insufflator-exsufflators if available) should be taught. [102]
Acute respiratory deteriorations due to infections require early management with antibiotics, chest physiotherapy and sometimes respiratory support. Advanced care directives regarding management of these situations should be discussed with the patient and their family from an early stage, providing information about the ventilatory and palliative options available. [102]
Nocturnal noninvasive intermittent positive pressure ventilation (NIPPV) is a safe and effective treatment for DMD patients with hypercapnia, with early initiation of NIPPV (before the development of daytime hypercapnia) an increasing trend. [14] Published consenses however, vary on specific recommendations for commencing NIPPV. [102],[103] In some studies nocturnal hypercapnia (PaCO 2 ≥ 45 mm Hg) alone has been used as a criterion to commence NIPPV, [104] whilst in others the presence of clinical symptoms is required. Benefits of NIPPV include increased quality of life, symptom relief, reduction and delay of onset of daytime hypercapnia, and improved life expectancy. [13],[15],[104],[105] Life expectancy has been documented to have increased to an average of 25 and even 30 years in patients who receive NIPPV.[105],[106],[107]
Further progression of respiratory failure requires fulltime ventilation, for example with 24 h nasal/mouthpiece ventilation, [108] or invasive mechanical ventilation via tracheostomy. These types of long-term ventilation are offered in some centers but the ethical, social and economic implications need to be discussed before this therapy is used.
Orthopedic issues
Maintenance of ambulation is foremost in the treatment of a child with DMD to prevent development of contractures and scoliosis, and to maintain independence.
In the early part of the disease course passive stretching, particularly of the Achilles tendons, iliotibial bands and hip flexors, and the use of night splints helps prevent development or progression of contractures. Despite these efforts, contractures ultimately develop and surgery is sometimes necessary.
Only prolongation of ambulation and corticosteroid therapy have been shown to delay the onset, and reduce the severity of scoliosis. [25],[109],[110] Surveillance radiographs for scoliosis should be performed yearly from age nine years. The average age of scoliosis surgery is 14-15 years. To qualify for surgery, the scoliosis should be greater than 25 degrees, with the vital capacity being greater than 30% of predicted. Baseline cardiac and respiratory function must be assessed prior. Benefits of scoliosis surgery include the prevention of gross deformity and discomfort, and perhaps some improvements to respiratory function. [111] It has not been shown to increase life expectancy. [112]
Maintenance of bone density is important in the prevention of fractures, particularly if the patient is on corticosteroid therapy. Monitoring of Vitamin D levels and supplementation of calcium and Vitamin D should be considered in all patients. [113],[114],[115]
General medical and psychosocial issues
Constipation is a common problem related to immobility and should be managed with an adequate bowel regime, high-fiber diet, and the use of laxatives. Behavioral issues can be a significant problem, particularly around the time of loss of ambulation and independence, and assistance from a psychologist or psychiatrist is sometimes required. Depression is probably under-diagnosed, and should be recognized and treated appropriately.
The provision of extra educational and physical assistance in the classroom is important, allowing many boys with DMD to attend a mainstream rather than a special educational school. This change in educational philosophy has been one of the major advances in management since the 1980s. Nevertheless there are still some children whose physical and intellectual deficits are such that they require a special educational setting. Some adolescents are able to proceed through the normal secondary education stream and undertake tertiary education. Obtaining meaningful employment remains a major issue.
Corticosteroid Therapy
Corticosteroids such as prednisolone, prednisone and deflazacort have been the only drugs shown to be effective to date in DMD. They should be offered to all patients, but only after a balanced discussion of the potential risks and benefits in the short and long term.
The specific mechanisms by which corticosteroids improve strength in DMD are not known, but various possibilities have been proposed. These include alteration of regulation of genes in muscle fibers, [116] slowing of the rate of skeletal muscle breakdown, [117] reducing cytotoxic T cells, [118] lowering cytosolic calcium concentrations, [119],[120] and increasing myogenic repair. [121]
A number of reviews and consensus statements have been published on the use of corticosteroids in DMD, the largest two being those published by the American Academy of Neurology (AAN), and the Cochrane database. [122],[123],[124],[125] The AAN practice parameter on the use of corticosteroids for the treatment of DMD [122] identified seven high-quality randomized controlled trials of prednisone/prednisolone, [126],[127],[128],[129],[130],[131],[132] and two of deflazacort. [133],[134] Results from these trials showed that prednisolone/prednisone improved muscle strength within 10 days, which was maximal at three months and maintained up to 18 months. Increases in muscle strength were paralleled by significant improvements in functional testing (e.g. time to arise from supine to standing, time to walk nine meters) and muscle mass, as measured by urinary creatinine excretion. [126]
The optimum dose of prednisolone/prednisone is felt to be 0.75 mg/kg/day. [122],[123] Lower doses of 0.3 mg/kg/day still result in improvements in strength and function, but to a lesser degree, [127],[128] but may be used if side-effects require a decrease in dose. Higher doses of 1.5 mg/kg/day did not result in additional benefits, [126] and alternate daily doses of 1.25 mg/kg and 2.5 mg/kg did not achieve the sustained benefits of daily dosing. [129] Other types of intermittent dosing such as prednisolone 10 days on and 10/20 days off, [135],[136] and twice weekly prednisolone (5 mg/kg/dose) [137] have also been described to be beneficial.
There are no good data on the optimal age to begin treatment with corticosteroids, or the optimal duration of treatment. [122] A common regimen is to offer corticosteroids at the time of decline of muscle strength and frequent falls, and to cease treatment when the child is no longer ambulant. Studies are lacking in the use of corticosteroids in very young children, and in wheelchair-bound patients. Use of intermittent corticosteroid regimens have been reported in children less than five years of age. [135],[138],[139]
Deflazacort, an oxazoline analogue of prednisone, is available in some countries as an alternative to prednisolone/prednisone. Treatment with deflazacort as a daily dose of 0.9 mg/kg/day produces similar sustained improvements in muscle strength and function [140],[141] although there are no large randomized controlled trials comparing deflazacort with prednisolone/prednisone.
There are no long-term randomized controlled trials that demonstrate that corticosteroids prolong the time to loss of ambulation in DMD or improve long-term survival, probably because of the large number of patients and long duration required of such a study to demonstrate these effects. [116] Non-randomized studies of long-term daily corticosteroids however, do suggest that ambulation may be prolonged by up to three to five years, [110],[142],[143],[144],[145] and that life expectancy is improved.[142]
Corticosteroids also appear to have a positive effect on the complications associated with DMD. Randomized and non-randomized studies have shown preservation of respiratory muscle strength and cough efficiency.[126],[127],[142],[146] Cardiac function is also better preserved, with a lower frequency of dilated cardiomyopathy seen in boys treated with corticosteroids. [142],[147],[148],[149] The prevalence and severity of scoliosis is also reduced in treated boys, with a subsequent delay and decrease in the need for spinal surgery. [110],[148],[150]
The commonest side-effects of corticosteroids are weight gain and development of a Cushingoid appearance, [122] with 75-80% of patients showing significant weight gain. [127],[128] Despite this, weight gain did not appear to adversely affect strength or function in these short-term studies. One small randomized trial suggests a lower incidence and severity of weight gain with deflazacort compared to prednisone. [140] Vertebral fractures have been detected in 32-40% of boys on long-term corticosteroids, although many are incidental and often not a reason to discontinue treatment. [110],[151] Long-bone fractures are also twice as likely compared to steroid-naοve patients. [110] Growth suppression is also seen after long-term corticosteroid treatment. [142] Cataracts (usually asymptomatic) appear to occur with high incidence in deflazacort-treated patients. [141],[142]
Other Drug Therapies
Oxandrolone, an anabolic steroid has shown some promise in increasing quantitative muscle strength in a randomized prospective trial, although not with sufficient magnitude to recommend it for routine use.[152]
Other immunosuppressive agents such as azathioprine and cyclosporine have also been studied. Azathioprine did not provide any benefit, [128] whilst cyclosporine showed some improvements in strength. [153] Creatine monohydrate has been associated with mild increases in muscle strength in DMD, but without significant improvements in functional measures. [154],[155],[156] Other drugs which have failed to show effect include nifedipine, [157] leucine, [158] selenium and Vitamin E, [159] and the antiserotinergic drugs methysergide [160] and pizotifen. [161]
Novel Therapies
Gene therapy
Viral vectors
The use of recombinant adeno-associated viral (rAAV) vectors that carry critical regions of the DMD gene is being explored, and is still in its early stages. [162] As only a small transcript size can be incorporated into available viral vectors, microdystrophin and minidystrophin genes have been created, with restoration of dystrophin expression demonstrated in mouse models. [163],[164] Challenges in this form of gene therapy include the potential need for immunosuppression, and optimizing delivery of the vectors to multiple muscle groups. [162] The use of non-viral plasmid vectors has also been described. [165],[166]
Antisense oligonucleotide exon skipping
Antisense oligonucleotides can be used to redirect splicing and induce exon skipping, with the aim of restoring the reading frame and producing a partially functioning dystrophin. This is ideal for the majority of DMD mutations, which are out-of-frame deletions or duplications. [167] Repeated intravenous administration of antisense oligonucleotides has successfully produced widespread dystrophin production in an mdx mouse model. [168] Successful exon skipping was recently demonstrated after intramuscular injection into the tibialis muscle in four patients. [169] Limitations to this form of therapy include ensuring sustained and safe beneficial effect of the oligonucleotide, as repeated lifelong administration would be required. In addition, different deletions will require different antisense oligonucleotides, making large-scale production difficult and costly. [170],[171]
Read-through stop codon strategies
Some aminoglycoside antibiotics such as gentamicin cause a relaxation in codon recognition, allowing read-through of nonsense mutations, which occur in around 7% of all DMD patients. [171] Read-through of the nonsense mutation in Exon 23 of the dystrophin gene in the mdx mouse model was demonstrated in 1999.[172] Subsequent clinical trials, however, have not been particularly successful. [173],[174]
PTC124 is a new orally administered investigative drug that promotes ribosomal read-through of stop codons, allowing continuation of translation and production of a functioning protein. [175] PTC124 has been shown to restore dystrophin levels in mdx mice, with an associated improvement in muscle function and decrease in CK levels. [176] Phase 1 studies in healthy adult volunteers showed that the drug is orally bioavailable and well tolerated. [177] A Phase 2 study of 26 boys demonstrated increased full-length dystrophin expression with PTC124, and decreased CK levels, but without significant improvement in timed functions or muscle strength. [178] Further clinical studies are underway. The main concern regarding the use of read-through stop codon drugs is the theoretical risk of a global read-through of physiological stop codons.
Stem cell therapy
Whilst early experiments of transplanted myoblasts showed promise, [179] subsequent studies showed that they were rejected by the host immune response in mdx mice and in patients with DMD. [180],[181] Autologous muscle-derived stem cells (myogenic stem cells) which are not at risk of host rejection can undergo ex vivo gene correction strategies such as small fragment homologous replacement and chimeraplasty, and be subsequently transplanted. [182],[183] Harvesting sufficient numbers of myogenic stem cells from dystrophic muscle however is a significant problem. [184],[185]
Bone marrow-derived stem cells have been shown to remodel muscle, and may be a renewable non-muscle cell type used to remodel dystrophic muscle. [186] However, because the number of stem cells with myoremodeling capacity is highly diluted when whole bone marrow is transplanted, strategies to rapidly isolate bone marrow-derived stem cells with myoremodeling capacity in sufficient numbers are required to make this a potential therapy.[187]
Utrophin
It is thought that utrophin, a protein homologue of dystrophin in the sarcolemma, may compensate for dystrophin deficiency if it is upregulated. [188] Various factors that increase utrophin expression are being explored, such as heregulin and L-arginine, but most are still in the very early stages. [189]
Prevention and genetic counseling
All families with an affected male with DMD should be referred for genetic counseling. Detection and counseling of female carriers is an important aspect of management for disease prevention. Prenatal diagnosis is available for carrier mothers, and is most accurate if a mutation is detected. Otherwise, linkage analysis is used. Germinal mosaicism is always a possibility in mothers who have negative molecular testing. The risk for future pregnancies being affected in such cases is generally given as 10-20%.
Carrier females of DMD should also be counseled about risks of cardiomyopathy. Periodic cardiovascular screening is recommended, commencing in early adulthood, [99],[190] with repeat evaluations at a minimum of every five years thereafter. [99]
Conclusions
Duchenne muscular dystrophy is a devastating condition that continues to affect many boys and their families. Recent advances in symptomatic management, with the careful use of corticosteroids, and respiratory support have increased life expectancy and quality of life considerably. The search for a cure remains elusive, although many promising and novel therapies are in progress, some of which have entered the stage of human trials.
References
| 1. | Monckton G, Hoskin V, Warren S. Prevalence and incidence of muscular dystrophy in Alberta, Canada. Clin Genet 1982;21:19-24. Back to cited text no. 1 [PUBMED] |
| 2. | Jones H, De Vivo DC, Darras BT. Neuromuscular disorders of infancy, childhood and adolescence. A clinician's approach. Oxford: Butterworth-Heinemann; 2003. Back to cited text no. 2 |
| 3. | Perloff JK. Cardiac rhythm and conduction in Duchenne's muscular dystrophy: A prospective study of 20 patients. J Am Coll Cardiol 1984;3:1263-8. Back to cited text no. 3 |
| 4. | Chenard AA, Becane HM, Tertrain F, de Kermadec JM, Weiss YA. Ventricular arrhythmia in Duchenne muscular dystrophy: Prevalence, significance and prognosis. Neuromuscul Disord 1993;3:201-6. Back to cited text no. 4 |
| 5. | Nigro G, Comi LI, Politano L, Bain RJ. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int J Cardiol 1990;26:271-7. Back to cited text no. 5 |
| 6. | Phillips MF, Quinlivan RC, Edwards RH, Calverley PM. Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am J Respir Crit Care Med 2001;164:2191-4. Back to cited text no. 6 |
| 7. | Rideau Y, Jankowski LW, Grellet J. Respiratory function in the muscular dystrophies. Muscle Nerve 1981;4:155-64. Back to cited text no. 7 |
| 8. | Baydur A, Gilgoff I, Prentice W, Carlson M, Fischer DA. Decline in respiratory function and experience with long-term assisted ventilation in advanced Duchenne's muscular dystrophy. Chest 1990;97:884-9. Back to cited text no. 8 |
| 9. | Inkley SR, Oldenburg FC, Vignos PJ Jr. Pulmonary function in Duchenne muscular dystrophy related to stage of disease. Am J Med 1974;56:297-306. Back to cited text no. 9 |
| 10. | Suresh S, Wales P, Dakin C, Harris MA, Cooper DG. Sleep-related breathing disorder in Duchenne muscular dystrophy: Disease spectrum in the paediatric population. J Paediatr Child Health 2005;41:500-3. Back to cited text no. 10 |
| 11. | Ragette R, Mellies U, Schwake C, Voit T, Teschler H. Patterns and predictors of sleep disordered breathing in primary myopathies. Thorax 2002;57:724-8. Back to cited text no. 11 |
| 12. | Hukins CA, Hillman DR. Daytime predictors of sleep hypoventilation in Duchenne muscular dystrophy. Am J Respir Crit Care Med 2000;161:166-70. Back to cited text no. 12 |
| 13. | Simonds AK, Muntoni F, Heather S, Fielding S. Impact of nasal ventilation on survival in hypercapnic Duchenne muscular dystrophy. Thorax 1998;53:949-52. Back to cited text no. 13 |
| 14. | Toussaint M, Chatwin M, Soudon P. Mechanical ventilation in Duchenne patients with chronic respiratory insufficiency: Clinical implications of 20 years published experience. Chron 2007;4:167-77. Back to cited text no. 14 |
| 15. | Vianello A, Bevilacqua M, Salvador V, Cardaioli C, Vincenti E. Long-term nasal intermittent positive pressure ventilation in advanced Duchenne's muscular dystrophy. Chest 1994;105:445-8. Back to cited text no. 15 |
| 16. | Leibowitz D, Dubowitz V. Intellect and behaviour in Duchenne muscular dystrophy. Dev Med Child Neurol 1981;23:577-90. Back to cited text no. 16 |
| 17. | Cotton S, Voudouris NJ, Greenwood KM. Intelligence and Duchenne muscular dystrophy: Full-scale, verbal and performance intelligence quotients. Dev Med Child Neurol 2001;43:497-501. Back to cited text no. 17 |
| 18. | Anderson JL, Head SI, Rae C, Morley JW. Brain function in Duchenne muscular dystrophy. Brain 2002;125:4-13. Back to cited text no. 18 |
| 19. | Karagan NJ, Richman LC, Sorensen JP. Analysis of verbal disability in Duchenne muscular dystrophy. J Nerv Ment Dis 1980;168:419-23. Back to cited text no. 19 |
| 20. | Karagan NJ. Intellectual functioning in Duchenne muscular dystrophy: A review. Psychol Bull 1979;86:250-9. Back to cited text no. 20 |
| 21. | Allen JE, Rodgin DW. Mental retardation in association with progressive dystrophy. Am J Dis Child 1960;100:208-11. Back to cited text no. 21 |
| 22. | Wu JY, Kuban KC, Allred E, Shapiro F, Darras BT. Association of Duchenne muscular dystrophy with autism spectrum disorder. J Child Neurol 2005;20:790-5. Back to cited text no. 22 |
| 23. | Hendriksen JG, Vles JS. Neuropsychiatric disorders in males with duchenne muscular dystrophy: Frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive--compulsive disorder. J Child Neurol 2008;23:477-81. Back to cited text no. 23 |
| 24. | Smith AD, Koreska J, Moseley CF. Progression of scoliosis in Duchenne muscular dystrophy. J Bone Joint Surg Am 1989;71:1066-74. Back to cited text no. 24 |
| 25. | Rodillo EB, Fernandez-Bermejo E, Heckmatt JZ, Dubowitz V. Prevention of rapidly progressive scoliosis in Duchenne muscular dystrophy by prolongation of walking with orthoses. J Child Neurol 1988;3:269-74. Back to cited text no. 25 |
| 26. | McDonald DG, Kinali M, Gallagher AC, Mercuri E, Muntoni F, Roper H, et al . Fracture prevalence in Duchenne muscular dystrophy. Dev Med Child Neurol 2002;44:695-8. Back to cited text no. 26 |
| 27. | Larson CM, Henderson RC. Bone mineral density and fractures in boys with Duchenne muscular dystrophy. J Pediatr Orthop 2000;20:71-4. Back to cited text no. 27 |
| 28. | Soderpalm AC, Magnusson P, Ahlander AC, Karlsson J, Kroksmark AK, Tulinius M, et al . Low bone mineral density and decreased bone turnover in Duchenne muscular dystrophy. Neuromuscul Disord 2007;17:919-28. Back to cited text no. 28 |
| 29. | Bianchi ML, Mazzanti A, Galbiati E, Saraifoger S, Dubini A, Cornelio F, . Bone mineral density and bone metabolism in Duchenne muscular dystrophy. Osteoporos Int 2003;14:761-7. Back to cited text no. 29 |
| 30. | Aparicio LF, Jurkovic M, DeLullo J. Decreased bone density in ambulatory patients with duchenne muscular dystrophy. J Pediatr Orthop 2002;22:179-81. Back to cited text no. 30 |
| 31. | Kelfer HM, Singer WD, Reynolds RN. Malignant hyperthermia in a child with Duchenne muscular dystrophy. Pediatrics 1983;71:118-9. Back to cited text no. 31 |
| 32. | Wedel DJ. Malignant hyperthermia and neuromuscular disease. Neuromuscul Disord 1992;2:157-64. Back to cited text no. 32 |
| 33. | Wang JM, Stanley TH. Duchenne muscular dystrophy and malignant hyperthermia-two case reports. Can Anaesth Soc J 1986;33:492-7. Back to cited text no. 33 |
| 34. | Hayes J, Veyckemans F, Bissonnette B. Duchenne muscular dystrophy: An old anesthesia problem revisited. Paediatr Anaesth 2008;18:100-6. Back to cited text no. 34 |
| 35. | Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkel LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987;50:509-17. Back to cited text no. 35 |
| 36. | Kunkel LM, Hejtmancik JF, Caskey CT, Speer A, Monaco AP, Middlesworth W, et al . Analysis of deletions in DNA from patients with Becker and Duchenne muscular dystrophy. Nature 1986;322:73-7. Back to cited text no. 36 |
| 37. | Kunkel LM, Beggs AH, Hoffman EP. Molecular genetics of Duchenne and Becker muscular dystrophy: Emphasis on improved diagnosis. Clin Chem 1989;35:B21-4. Back to cited text no. 37 |
| 38. | Mandel JL. Dystrophin. The gene and its product. Nature 1989;339:584-6. Back to cited text no. 38 |
| 39. | Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987;51:919-28. Back to cited text no. 39 |
| 40. | Zubrzycka-Gaarn EE, Bulman DE, Karpati G, Burghes AH, Belfall B, Klamut HJ, et al . The Duchenne muscular dystrophy gene product is localized in sarcolemma of human skeletal muscle. Nature 1988;333:466-9. Back to cited text no. 40 |
| 41. | Ervasti JM, Ohlendieck K, Kahl SD, Gaver MG, Campbell KP. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990;345:315-9. Back to cited text no. 41 |
| 42. | Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve 2001;24:1575-94. Back to cited text no. 42 |
| 43. | Koenig M, Monaco AP, Kunkel LM. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988;53:219-28. Back to cited text no. 43 |
| 44. | Rentschler S, Linn H, Deininger K, Bedford MT, Espanel X, Sudol M. The WW domain of dystrophin requires EF-hands region to interact with beta-dystroglycan. Biol Chem 1999;380:431-42. Back to cited text no. 44 |
| 45. | Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature 1989;338:259-62. Back to cited text no. 45 |
| 46. | Ahn AH, Kunkel LM. The structural and functional diversity of dystrophin. Nat Genet 1993;3:283-91. Back to cited text no. 46 |
| 47. | Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988;2:90-5. Back to cited text no. 47 |
| 48. | Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006;34:135-44. Back to cited text no. 48 |
| 49. | Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet neurol 2003;2:731-40. Back to cited text no. 49 |
| 50. | Beggs AH, Koenig M, Boyce FM, Kunkel LM. Detection of 98% of DMD/BMD gene deletions by polymerase chain reaction. Hum Genet 1990;86:45-8. Back to cited text no. 50 |
| 51. | Liechti-Gallati S, Koenig M, Kunkel LM, Frey D, Boltshauser E, Schneider V, et al . Molecular deletion patterns in Duchenne and Becker type muscular dystrophy. Hum Genet 1989;81:343-8. Back to cited text no. 51 |
| 52. | White SJ, Aartsma-Rus A, Flanigan KM, Weiss RB, Kneppers ALJ, Lalic T, et al . Duplications in the DMD gene. Hum Mutat 2006;27:938-45. Back to cited text no. 52 |
| 53. | Roberts RG, Gardner RJ, Bobrow M. Searching for the 1 in 2,400,000: A review of dystrophin gene point mutations. Hum Mutat 1994;4:1-11. Back to cited text no. 53 |
| 54. | Petrof BJ. The molecular basis of activity-induced muscle injury in Duchenne muscular dystrophy. Mol Cell Biochem 1998;179:111-23. Back to cited text no. 54 |
| 55. | Deconinck N, Dan B. Pathophysiology of duchenne muscular dystrophy: Current hypotheses. Pediatr Neurol 2007;36:1-7. Back to cited text no. 55 |
| 56. | Morandi L, Mora M, Gussoni E, Tedeschi S, Cornelio F. Dystrophin analysis in Duchenne and Becker muscular dystrophy carriers: Correlation with intracellular calcium and albumin. Ann Neurol 1990;28:674-9. Back to cited text no. 56 |
| 57. | Bodensteiner JB, Engel AG. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: A study of 567,000 muscle fibers in 114 biopsies. Neurology 1978;28:439-46. Back to cited text no. 57 |
| 58. | Imbert N, Vandebrouck C, Duport G, Raymond G, Hassoni AA, Constantin B, et al . Calcium currents and transients in co-cultured contracting normal and Duchenne muscular dystrophy human myotubes. J Physiol 2001;534:343-55. Back to cited text no. 58 |
| 59. | Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995;82:743-52. Back to cited text no. 59 |
| 60. | Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, et al . Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA 2000;97:13818-23. Back to cited text no. 60 |
| 61. | Crosbie RH, Straub V, Yun HY, Lee JC, Rafael JA, Chamberlain JS, et al . MDX muscle pathology is independent of nNOS perturbation. Hum Mol Genet 1998;7:823-9. Back to cited text no. 61 |
| 62. | Kapsa R, Kornberg AJ, Byrne E. Novel therapies for Duchenne muscular dystrophy. Lancet Neurol 2003;2:299-310. Back to cited text no. 62 |
| 63. | Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the MDX mouse: A point mutation. Science 1989;244:1578-80. Back to cited text no. 63 |
| 64. | Rosalki SB. Serum enzymes in disease of skeletal muscle. Clin Lab Med 1989;9:767-81. Back to cited text no. 64 |
| 65. | Brooke MH, Fenichel GM, Griggs RC, Mendell JR, Moxley R, Miller JP, et al . Clinical investigation in Duchenne dystrophy: 2, Determination of the "power" of therapeutic trials based on the natural history. Muscle Nerve 1983;6:91-103. Back to cited text no. 65 |
| 66. | Zatz M, Rapaport D, Vainzof M, Passos-Bueno MR, Bortolini ER, Pavanello Rde C, et al . Serum creatine-kinase (CK) and pyruvate-kinase (PK) activities in Duchenne (DMD) as compared with Becker (BMD) muscular dystrophy. J Neurol Sci 1991;102:190-6. Back to cited text no. 66 |
| 67. | Tay SK, Ong HT, Low PS. Transaminitis in Duchenne's muscular dystrophy. Ann Acad Med Singapore 2000;29:719-22. Back to cited text no. 67 |
| 68. | Morse RP, Rosman NP. Diagnosis of occult muscular dystrophy: Importance of the "chance" finding of elevated serum aminotransferase activities. J Pediatr 1993;122:254-6. Back to cited text no. 68 |
| 69. | Munsat TL, Baloh R, Pearson CM, Fowler W Jr. Serum enzyme alterations in neuromuscular disorders. JAMA 1973;226:1536-43. Back to cited text no. 69 |
| 70. | Rowland LP. Biochemistry of muscle membranes in Duchenne muscular dystrophy. Muscle Nerve 1980;3:3-20. Back to cited text no. 70 |
| 71. | Korones DN, Brown MR, Palis J. "Liver function tests" are not always tests of liver function. Am J Hematol 2001;66:46-8. Back to cited text no. 71 |
| 72. | Lott JA, Landesman PW. The enzymology of skeletal muscle disorders. Crit Rev Clin Lab Sci 1984;20:153-90. Back to cited text no. 72 |
| 73. | Desmedt JE, Borenstein S. Relationship of spontaneous fibrillation potentials to muscle fibre segmentation in human muscular dystrophy. Nature 1975;258:531-4. Back to cited text no. 73 |
| 74. | Desmedt JE, Borenstein S. Regeneration in Duchenne muscular dystrophy: Electromyographic evidence. Arch Neurol 1976;33:642-50. Back to cited text no. 74 |
| 75. | Muntoni F. Is a muscle biopsy in Duchenne dystrophy really necessary? Neurology 2001;57:574-5. Back to cited text no. 75 |
| 76. | Bell CD, Conen PE. Change in fiber size in Duchenne muscular dystrophy. Neurology 1967;17:902-13. Back to cited text no. 76 |
| 77. | Bulman DE, Murphy EG, Zubrzycka-Gaarn EE, Worton RG, Ray PN. Differentiation of Duchenne and Becker muscular dystrophy phenotypes with amino and carboxy-terminal antisera specific for dystrophin. Am J Hum Genet 1991;48:295-304. Back to cited text no. 77 |
| 78. | Voit T, Stuettgen P, Cremer M, Goebel HH. Dystrophin as a diagnostic marker in Duchenne and Becker muscular dystrophy: Correlation of immunofluorescence and western blot. Neuropediatrics 1991;22:152-62. Back to cited text no. 78 |
| 79. | Nicholson LV, Johnson MA, Gardner-Medwin D, Bhattacharya S, Harris JB. Heterogeneity of dystrophin expression in patients with Duchenne and Becker muscular dystrophy. Acta Neuropathol 1990;80:239-50. Back to cited text no. 79 |
| 80. | Nicholson LV, Johnson MA, Bushby KM, Gardner-Medwin D, Curtis A, Ginjaar IB, et al . Integrated study of 100 patients with Xp21 linked muscular dystrophy using clinical, genetic, immunochemical, and histopathological data: Part 1, Trends across the clinical groups. J Med Genet 1993;30:728-36. Back to cited text no. 80 |
| 81. | Hoffman EP, Fischbeck KH, Brown RH, Johnson M, Medori R, Loike JD, et al . Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N Engl J Med 1988;318:1363-8. Back to cited text no. 81 |
| 82. | Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res 1988;16:11141-56. Back to cited text no. 82 |
| 83. | Schwartz M, Duno M. Improved molecular diagnosis of dystrophin gene mutations using the multiplex ligation-dependent probe amplification method. Genet Test 2004;8:361-7. Back to cited text no. 83 |
| 84. | Lalic T, Vossen RH, Coffa J, Schouten JP, Guc-Scekic M, Radivojevic D, et al . Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet 2005;13:1231-4. Back to cited text no. 84 |
| 85. | Janssen B, Hartmann C, Scholz V, Jauch A, Zschocke J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: Potential and pitfalls. Neurogenetics 2005;6:29-35. Back to cited text no. 85 |
| 86. | Gatta V, Scarciolla O, Gaspari AR, Palka C, De Angelis MV, Di Muzio A, et al . Identification of deletions and duplications of the DMD gene in affected males and carrier females by multiple ligation probe amplification (MLPA). Hum Genet 2005;117:92-8. Back to cited text no. 86 |
| 87. | Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet 2003;72:931-9. Back to cited text no. 87 |
| 88. | Bennett RR, den Dunnen J, O'Brien KF, Darras BT, Kunkel LM. Detection of mutations in the dystrophin gene via automated DHPLC screening and direct sequencing. BMC Genet 2001;2:17. Back to cited text no. 88 |
| 89. | Hegde MR, Chin EL, Mulle JG, Okou DT, Warren ST, Zwick ME. Microarray-based mutation detection in the dystrophin gene. Hum Mutat 2008;29:1091-9. Back to cited text no. 89 |
| 90. | Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited neuromuscular disorders: Past, present, and future. J Magn Reson Imaging 2007;25:433-40. Back to cited text no. 90 |
| 91. | Hoogerwaard EM, Bakker E, Ippel PF, Oosterwijk JC, Majoor-Krakauer DF, Leschot NJ, et al . Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in The Netherlands: A cohort study. Lancet 1999;353:2116-9. Back to cited text no. 91 |
| 92. | Norman A, Harper P. A survey of manifesting carriers of Duchenne and Becker muscular dystrophy in Wales. Clin Genet 1989;36:31-7. Back to cited text no. 92 |
| 93. | Hoogerwaard EM, van der Wouw PA, Wilde AA, Bakker E, Ippel PF, Oosterwijk JC, et al . Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 1999;9:347-51. Back to cited text no. 93 |
| 94. | Melacini P, Fanin M, Angelini A, Pegoraro E, Livi U, Danieli GA, et al . Cardiac transplantation in a Duchenne muscular dystrophy carrier. Neuromuscul Disord 1998;8:585-90. Back to cited text no. 94 |
| 95. | Holloway SM, Wilcox DE, Wilcox A, Dean JC, Berg JN, Goudie DR, et al . Life expectancy and death from cardiomyopathy amongst carriers of Duchenne and Becker muscular dystrophy in Scotland. Heart 2008;94:633-6. Back to cited text no. 95 |
| 96. | Politano L, Nigro V, Nigro G, Petretta VR, Passamano L, Papparella S, et al . Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA 1996;275:1335-8. Back to cited text no. 96 |
| 97. | Arahata K, Ishihara T, Kamakura K, Tsukahara T, Ishiura S, Baba C, et al . Mosaic expression of dystrophin in symptomatic carriers of Duchenne's muscular dystrophy. N Engl J Med 1989;320:138-42. Back to cited text no. 97 |
| 98. | Hoogerwaard EM, Ginjaar IB, Bakker E, de Visser M. Dystrophin analysis in carriers of Duchenne and Becker muscular dystrophy. Neurology 2005;65:1984-6. Back to cited text no. 98 |
| 99. | American Academy of Pediatrics Section on C, Cardiac S. Cardiovascular health supervision for individuals affected by Duchenne or Becker muscular dystrophy. Pediatrics 2005;116:1569-73. Back to cited text no. 99 |
| 100. | Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, et al . Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation 2005;112:2799-804. Back to cited text no. 100 |
| 101. | McNally EM, MacLeod H. Therapy insight: Cardiovascular complications associated with muscular dystrophies. Nat Clin Pract Cardiovasc Med 2005;2:301-8. Back to cited text no. 101 |
| 102. | Finder JD, Birnkrant D, Carl J, Farber HJ, Gozal D, Iannaccone ST, et al . Respiratory care of the patient with Duchenne muscular dystrophy: ATS consensus statement. Am J Respir Crit Care Med 2004;170:456-65. Back to cited text no. 102 |
| 103. | Robert D, Willig TN, Leger P, Paulus J. Long-term nasal ventilation in neuromuscular disorders: Report of a consensus conference. Eur Respir J 1993;6:599-606. Back to cited text no. 103 |
| 104. | Ward S, Chatwin M, Heather S, Simonds AK. Randomized controlled trial of non-invasive ventilation (NIV) for nocturnal hypoventilation in neuromuscular and chest wall disease patients with daytime normocapnia. Thorax 2005;60:1019-24. Back to cited text no. 104 |
| 105. | Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord 2002;12:926-9. Back to cited text no. 105 |
| 106. | Eagle M, Bourke J, Bullock R, Gibson M, Mehta J, Giddings D, et al . Managing Duchenne muscular dystrophy--the additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord 2007;17:470-5. Back to cited text no. 106 |
| 107. | Konagaya M, Sakai M, Wakayama T, Kimura S, Kuru S, Yasuma F. Effect of intermittent positive pressure ventilation on life-span and causes of death in Duchenne muscular dystrophy. Rinsho Shinkeigaku 2005;45:643-6. Back to cited text no. 107 |
| 108. | Toussaint M, Steens M, Wasteels G, Soudon P. Diurnal ventilation via mouthpiece: Survival in end-stage Duchenne patients. Eur Respir J 2006;28:549-55. Back to cited text no. 108 |
| 109. | Kinali M, Main M, Eliahoo J, Messina S, Knight RK, Lehovsky J, et al . Predictive factors for the development of scoliosis in Duchenne muscular dystrophy. Eur J Paediatr Neurol 2007;11:160-6. Back to cited text no. 109 |
| 110. | King WM, Ruttencutter R, Nagaraja HN, Matkovic V, Landoll J, Hoyle C, et al . Orthopedic outcomes of long-term daily corticosteroid treatment in Duchenne muscular dystrophy. Neurology 2007;68:1607-13. Back to cited text no. 110 |
| 111. | Velasco MV, Colin AA, Zurakowski D, Darras BT, Shapiro F. Posterior spinal fusion for scoliosis in duchenne muscular dystrophy diminishes the rate of respiratory decline. Spine 2007;32:459-65. Back to cited text no. 111 |
| 112. | Cheuk DK, Wong V, Wraige E, Baxter P, Cole A, N'Diaye T, et al . Surgery for scoliosis in Duchenne muscular dystrophy. Cochrane Database Syst Rev 2007;1:CD005375. Back to cited text no. 112 |
| 113. | Bachrach LK. Taking steps towards reducing osteoporosis in Duchenne muscular dystrophy. Neuromuscul Disord 2005;15:86-7. Back to cited text no. 113 |
| 114. | Biggar WD, Bachrach LK, Henderson RC, Kalkwarf H, Plotkin H, Wong BL. Bone health in Duchenne muscular dystrophy: A workshop report from the meeting in Cincinnati, Ohio, July 8, 2004. Neuromuscul Disord 2005;15:80-5. Back to cited text no. 114 |
| 115. | Quinlivan R, Roper H, Davie M, Shaw NJ, McDonagh J, Bushby K. Report of a Muscular Dystrophy Campaign funded workshop Birmingham, UK, January 16th 2004, Osteoporosis in Duchenne muscular dystrophy: Its prevalence, treatment and prevention. Neuromuscul Disord 2005;15:72-9. Back to cited text no. 115 |
| 116. | Muntoni F, Fisher I, Morgan JE, Abraham D. Steroids in Duchenne muscular dystrophy: From clinical trials to genomic research. Neuromuscul Disord 2002;12:S162-5. Back to cited text no. 116 |
| 117. | Rifai Z, Welle S, Moxley RT 3rd, Lorenson M, Griggs RC. Effect of prednisone on protein metabolism in Duchenne dystrophy. Am J Physiol 1995;268:E67-74. Back to cited text no. 117 |
| 118. | Kissel JT, Burrow KL, Rammohan KW, Mendell JR. Mononuclear cell analysis of muscle biopsies in prednisone-treated and untreated Duchenne muscular dystrophy: CIDD Study Group. Neurology 1991;41:667-72. Back to cited text no. 118 |
| 119. | Metzinger L, Passaquin AC, Leijendekker WJ, Poindron P, Ruegg UT. Modulation by prednisolone of calcium handling in skeletal muscle cells. Br J Pharmacol 1995;116:2811-6. Back to cited text no. 119 |
| 120. | Passaquin AC, Lhote P, Ruegg UT. Calcium influx inhibition by steroids and analogs in C2C12 skeletal muscle cells. Br J Pharmacol 1998;124:1751-9. Back to cited text no. 120 |
| 121. | Anderson JE, Weber M, Vargas C. Deflazacort increases laminin expression and myogenic repair, and induces early persistent functional gain in mdx mouse muscular dystrophy. Cell Transplant 2000;9:551-64. Back to cited text no. 121 |
| 122. | Moxley RT, 3rd, Ashwal S, Pandya S, Connolly A, Florence J, Mathews K, et al . Practice parameter: Corticosteroid treatment of Duchenne dystrophy: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2005;64:13-20. Back to cited text no. 122 |
| 123. | Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy.[update of Cochrane Database Syst Rev. 2004;(2):CD003725; PMID:15106215]. Cochrane Database Syst Rev 2008:CD003725. Back to cited text no. 123 |
| 124. | Bushby K, Muntoni F, Urtizberea A, Hughes R, Griggs R. Report on the 124th ENMC International Workshop: Treatment of Duchenne muscular dystrophy: Defining the gold standards of management in the use of corticosteroids. 2-4 April 2004, Naarden, The Netherlands. Neuromuscul Disord 2004;14:526-34. Back to cited text no. 124 |
| 125. | Wong BL, Christopher C. Corticosteroids in Duchenne muscular dystrophy: A reappraisal. J Child Neurol 2002;17:183-90. Back to cited text no. 125 |
| 126. | Mendell JR, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, Miller JP, et al . Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N Engl J Med 1989;320:1592-7. Back to cited text no. 126 |
| 127. | Griggs RC, Moxley RT 3rd, Mendell JR, Fenichel GM, Brooke MH, Pestronk A, et al . Prednisone in Duchenne dystrophy: A randomized, controlled trial defining the time course and dose response. Arch Neurol 1991;48:383-8. Back to cited text no. 127 |
| 128. | Griggs RC, Moxley RT 3rd, Mendell JR, Fenichel GM, Brooke MH, Pestronk A, et al . Duchenne dystrophy: Randomized, controlled trial of prednisone (18 months) and azathioprine (12 months). Neurology 1993;43:520-7. Back to cited text no. 128 |
| 129. | Fenichel GM, Mendell JR, Moxley RT 3rd, Griggs RC, Brooke MH, Miller JP, et al . A comparison of daily and alternate-day prednisone therapy in the treatment of Duchenne muscular dystrophy. Arch Neurol 1991;48:575-9. Back to cited text no. 129 |
| 130. | Rahman MM, Hannan MA, Mondol BA, Bhoumick NB, Haque A. Prednisolone in Duchenne muscular dystrophy. Bangladesh Med Res Counc Bull 2001;27:38-42. Back to cited text no. 130 |
| 131. | Backman E, Henriksson KG. Low-dose prednisolone treatment in Duchenne and Becker muscular dystrophy. Neuromuscul Disord 1995;5:233-41. Back to cited text no. 131 |
| 132. | Siegel IM, Miller JE, Ray RD. Failure of corticosteroid in the treatment of Duchenne (pseudo-hypertrophic) muscular dystrophy: Report of a clinically matched three year double-blind study. IMJ Ill Med J 1974;145:32-33 passim. Back to cited text no. 132 |
| 133. | Mesa LE, Dubrovsky AL, Corderi J, Marco P, Flores D. Steroids in Duchenne muscular dystrophy--deflazacort trial. Neuromuscul Disord 1991;1:261-6. Back to cited text no. 133 |
| 134. | Angelini C, Pegoraro E, Turella E, Intino MT, Pini A, Costa C. Deflazacort in Duchenne dystrophy: Study of long-term effect. Muscle Nerve 1994;17:386-91. Back to cited text no. 134 |
| 135. | Kinali M, Mercuri E, Main M, Muntoni F, Dubowitz V. An effective, low-dosage, intermittent schedule of prednisolone in the long-term treatment of early cases of Duchenne dystrophy. Neuromuscul Disord 2002;12:S169-74. Back to cited text no. 135 |
| 136. | Dubowitz V. Prednisone in Duchenne dystrophy. Neuromuscul Disord 1991;1:161-3. Back to cited text no. 136 |
| 137. | Connolly AM, Schierbecker J, Renna R, Florence J. High dose weekly oral prednisone improves strength in boys with Duchenne muscular dystrophy. Neuromuscul Disord 2002;12:917-25. Back to cited text no. 137 |
| 138. | Dubowitz V, Kinali M, Main M, Mercuri E, Muntoni F. Remission of clinical signs in early duchenne muscular dystrophy on intermittent low-dosage prednisolone therapy. Eur J Paediatr Neurol 2002;6:153-9. Back to cited text no. 138 |
| 139. | Merlini L, Cicognani A, Malaspina E, Gennari M, Gnudi S, Talim B, et al . Early prednisone treatment in Duchenne muscular dystrophy. Muscle Nerve 2003;27:222-7. Back to cited text no. 139 |
| 140. | Bonifati MD, Ruzza G, Bonometto P, Berardinelli A, Gorni K, Orcesi S, et al . A multicenter, double-blind, randomized trial of deflazacort versus prednisone in Duchenne muscular dystrophy. Muscle Nerve 2000;23:1344-7. Back to cited text no. 140 |
| 141. | Reitter B. Deflazacort vs. prednisone in Duchenne muscular dystrophy: Trends of an ongoing study. Brain Dev 1995;17:39-43. Back to cited text no. 141 |
| 142. | Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord 2006;16:249-55. Back to cited text no. 142 |
| 143. | Biggar WD, Gingras M, Fehlings DL, Harris VA, Steele CA. Deflazacort treatment of Duchenne muscular dystrophy. J Pediatr 2001;138:45-50. Back to cited text no. 143 |
| 144. | Balaban B, Matthews DJ, Clayton GH, Carry T. Corticosteroid treatment and functional improvement in Duchenne muscular dystrophy: Long-term effect. Am J Phys Med Rehabil 2005;84:843-50. Back to cited text no. 144 |
| 145. | Pandya S, Myers G, Moxley R. Effect of daily prednisone on independent ambulation in patients with Duchenne dystrophy treated for up to 15 years. Neuromuscul Disord 2001;11:630. Back to cited text no. 145 |
| 146. | Daftary AS, Crisanti M, Kalra M, Wong B, Amin R. Effect of long-term steroids on cough efficiency and respiratory muscle strength in patients with Duchenne muscular dystrophy. Pediatrics 2007;119:e320-4. Back to cited text no. 146 |
| 147. | Markham LW, Spicer RL, Khoury PR, Wong BL, Mathews KD, Cripe LH. Steroid therapy and cardiac function in Duchenne muscular dystrophy. Pediatr Cardiol 2005;26:768-71. Back to cited text no. 147 |
| 148. | Houde S, Filiatrault M, Fournier A, Dube J, D'Arcy S, Berube D, et al . Deflazacort use in Duchenne muscular dystrophy: An 8-year follow-up. Pediatr Neurol 2008;38:200-6. Back to cited text no. 148 |
| 149. | Silversides CK, Webb GD, Harris VA, Biggar DW. Effects of deflazacort on left ventricular function in patients with Duchenne muscular dystrophy. Am J Cardiol 2003;91:769-72. Back to cited text no. 149 |
| 150. | Alman BA, Raza SN, Biggar WD. Steroid treatment and the development of scoliosis in males with duchenne muscular dystrophy. J Bone Joint Surg Am 2004;86:519-24. Back to cited text no. 150 |
| 151. | Bothwell JE, Gordon KE, Dooley JM, MacSween J, Cummings EA, Salisbury S. Vertebral fractures in boys with Duchenne muscular dystrophy. Clin Pediatr (Phila) 2003;42:353-6. Back to cited text no. 151 |
| 152. | Fenichel GM, Griggs RC, Kissel J, Kramer TI, Mendell JR, Moxley RT, et al . A randomized efficacy and safety trial of oxandrolone in the treatment of Duchenne dystrophy. Neurology 2001;56:1075-9. Back to cited text no. 152 |
| 153. | Sharma KR, Mynhier MA, Miller RG. Cyclosporine increases muscular force generation in Duchenne muscular dystrophy. Neurology 1993;43:527-32. Back to cited text no. 153 |
| 154. | Walter MC, Lochmuller H, Reilich P, Klopstock T, Huber R, Hartard M, et al . Creatine monohydrate in muscular dystrophies: A double-blind, placebo-controlled clinical study. Neurology 2000;54:1848-50. Back to cited text no. 154 |
| 155. | Kley RA, Vorgerd M, Tarnopolsky MA. Creatine for treating muscle disorders. Cochrane Database Syst Rev 2007;1:CD004760. Back to cited text no. 155 |
| 156. | Tarnopolsky MA, Mahoney DJ, Vajsar J, Rodriguez C, Doherty TJ, Roy BD, et al . Creatine monohydrate enhances strength and body composition in Duchenne muscular dystrophy. Neurology 2004;62:1771-7. Back to cited text no. 156 |
| 157. | Moxley RT 3rd, Brooke MH, Fenichel GM, Mendell JR, Griggs RC, Miller JP, et al . Clinical investigation in Duchenne dystrophy. VI: Double-blind controlled trial of nifedipine. Muscle Nerve 1987;10:22-33. Back to cited text no. 157 |
| 158. | Mendell JR, Griggs RC, Moxley RT 3rd, Fenichel GM, Brooke MH, Miller JP, et al . Clinical investigation in Duchenne muscular dystrophy, IV: Double-blind controlled trial of leucine. Muscle Nerve 1984;7:535-541. Back to cited text no. 158 |
| 159. | Gamstorp I, Gustavson KH, Hellstrom O, Nordgren B. A trial of selenium and vitamin E in boys with muscular dystrophy. J Child Neurol 1986;1:211-4. Back to cited text no. 159 |
| 160. | Patten BM, Zeller RS. Clinical trials of vasoactive and antiserotonin drugs in Duchenne muscular dystrophy. Ann Clin Res 1983;15:164-6. Back to cited text no. 160 |
| 161. | Steru D, Paclet JP, Barthelet G, Gailliard G, Piton A, Monchartre E, et al . Double-blind study of the efficacy of an antiserotoninergic drug, pizotifen, in Duchenne's muscular dystrophy. Arch Fr Pediatr 1987;44:461-5. Back to cited text no. 161 |
| 162. | Rodino-Klapac LR, Chicoine LG, Kaspar BK, Mendell JR. Gene therapy for duchenne muscular dystrophy: Expectations and challenges. Arch Neurol 2007;64:1236-41. Back to cited text no. 162 |
| 163. | Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al . rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med 2006;12:787-9. Back to cited text no. 163 |
| 164. | Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in MDX mouse model. Proc Natl Acad Sci USA 2000;97:13714-9. Back to cited text no. 164 |
| 165. | Romero NB, Braun S, Benveniste O, Leturcq F, Hogrel JY, Morris GE, et al . Phase I study of dystrophin plasmid-based gene therapy in Duchenne/Becker muscular dystrophy. Hum Gene Ther 2004;15:1065-76. Back to cited text no. 165 |
| 166. | Rando TA. Non-viral gene therapy for Duchenne muscular dystrophy: Progress and challenges. Biochim Biophys Acta 2007;1772:263-71. Back to cited text no. 166 |
| 167. | Wilton SD, Fletcher S. Antisense oligonucleotides, exon skipping and the dystrophin gene transcript. Acta Myol 2005;24:222-9. Back to cited text no. 167 |
| 168. | Alter J, Lou F, Rabinowitz A, Yin H, Rosenfeld J, Wilton SD, et al . Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med 2006;12:175-7. Back to cited text no. 168 |
| 169. | van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M, et al . Local dystrophin restoration with antisense oligonucleotide PRO051. N Engl J Med 2007;357:2677-86. Back to cited text no. 169 |
| 170. | Bertoni C. Clinical approaches in the treatment of Duchenne muscular dystrophy (DMD) using oligonucleotides. Front Biosci 2008;13:517-27. Back to cited text no. 170 |
| 171. | Muntoni F, Wells D. Genetic treatments in muscular dystrophies. Curr Opin Neurol 2007;20:590-4. Back to cited text no. 171 |
| 172. | Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest 1999;104:375-81. Back to cited text no. 172 |
| 173. | Politano L, Nigro G, Nigro V, Piluso G, Papparella S, Paciello O, et al . Gentamicin administration in Duchenne patients with premature stop codon: Preliminary results. Acta Myol 2003;22:15-21. Back to cited text no. 173 |
| 174. | Wagner KR, Hamed S, Hadley DW, Gropman AL, Burstein AH, Escolar DM, et al . Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol 2001;49:706-11. Back to cited text no. 174 |
| 175. | Hamed SA. Drug evaluation: PTC-124--a potential treatment of cystic fibrosis and Duchenne muscular dystrophy. IDrugs 2006;9:783-9. Back to cited text no. 175 |
| 176. | Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al . PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007;447:87-91. Back to cited text no. 176 |
| 177. | Hirawat S, Welch EM, Elfring GL, Northcutt VJ, Paushkin S, Hwang S, et al . Safety, tolerability, and pharmacokinetics of PTC124: A nonaminoglycoside nonsense mutation suppressor, following single- and multiple-dose administration to healthy male and female adult volunteers. J Clin Pharmacol 2007;47:430-44. Back to cited text no. 177 |
| 178. | Wong B, Bonnemann C, Finkel R, Flanigan KM, Sampson J, Sweeney L, et al . Phase 2 study of PTC124 for nonsense mutation suppression therapy of Duchenne Muscular Dystrophy (DMD). Ann Neurol 2007;62:S96. Back to cited text no. 178 |
| 179. | Partridge TA, Morgan JE, Coulton GR, Hoffman EP, Kunkel LM. Conversion of mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature 1989;337:176-9. Back to cited text no. 179 |
| 180. | Tremblay JP, Malouin F, Roy R, Huard J, Bouchard JP, Satoh A, et al . Results of a triple blind clinical study of myoblast transplantations without immunosuppressive treatment in young boys with Duchenne muscular dystrophy. Cell Transplant 1993;2:99-112. Back to cited text no. 180 |
| 181. | Huard J, Roy R, Guerette B, Verreault S, Tremblay G, Tremblay JP. Human myoblast transplantation in immunodeficient and immunosuppressed mice: Evidence of rejection. Muscle Nerve 1994;17:224-34. Back to cited text no. 181 |
| 182. | Kapsa R, Quigley A, Lynch GS, Steeper K, Kornberg AJ, Gregorevic P, et al . In vivo and in vitro correction of the mdx dystrophin gene nonsense mutation by short-fragment homologous replacement. Hum Gene Ther 2001;12:629-42. Back to cited text no. 182 |
| 183. | Bertoni C, Rando TA. Dystrophin gene repair in mdx muscle precursor cells in vitro and in vivo mediated by RNA-DNA chimeric oligonucleotides. Hum Gene Ther 2002;13:707-18. Back to cited text no. 183 |
| 184. | Cossu G, Mavilio F. Myogenic stem cells for the therapy of primary myopathies: Wishful thinking or therapeutic perspective? J Clin Invest 2000;105:1669-74. Back to cited text no. 184 |
| 185. | Blau HM, Webster C, Pavlath GK. Defective myoblasts identified in Duchenne muscular dystrophy. Proc Natl Acad Sci USA 1983;80:4856-60. Back to cited text no. 185 |
| 186. | Ferrari G, Cusella-De Angelis G, Coletta M, Paolucci E, Stornaiuolo A, Cossu G, et al . Muscle regeneration by bone marrow-derived myogenic progenitors. Science 1998;279:1528-30. Back to cited text no. 186 |
| 187. | Wong SH, Lowes KN, Bertoncello I, Quigley AF, Simmons PJ, Cook MJ, et al . Evaluation of Sca-1 and c-Kit as selective markers for muscle remodelling by nonhemopoietic bone marrow cells. Stem Cells 2007;25:1364-74. Back to cited text no. 187 |
| 188. | Clerk A, Morris GE, Dubowitz V, Davies KE, Sewry CA. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem J 1993;25:554-61. Back to cited text no. 188 |
| 189. | Miura P, Jasmin BJ. Utrophin upregulation for treating Duchenne or Becker muscular dystrophy: How close are we? Trends Mol Med 2006;12:122-9. Back to cited text no. 189 |
| 190. | Nolan MA, Jones OD, Pedersen RL, Johnston HM. Cardiac assessment in childhood carriers of Duchenne and Becker muscular dystrophies. Neuromuscul Disord 2003;13:129-32. Back to cited text no. 190 |
Copyright 2008 - Neurology India
The following images related to this document are available:
Photo images
[ni08073f2.jpg]
[ni08073f1.jpg]
[ni08073f3.jpg]
|

{kind=link}
{kind=link}
{kind=link}