
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 56, Num. 3, 2008, pp. 263-270
|
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 263-270
Review Article
Inflammatory muscle diseases
Mastaglia FL
Centre for Neuromuscular and Neurological Disorders, University of Western Australia, Queen Elizabeth II Medical Centre, Nedlands, 6009
Correspondence Address:Centre for Neuromuscular and Neurological Disorders, University of Western Australia, Queen Elizabeth II Medical Centre, Nedlands, 6009
flmast@cyllene.uwa.edu.au
Date of Acceptance: 23-Jul-2008
Code Number: ni08076
Abstract The three major immune-mediated inflammatory myopathies, dermatomyositis (DM), polymyositis (PM) and inclusion body myositis (IBM), each have their own distinctive clinical features, underlying pathogenetic mechanisms and patterns of muscle gene expression. In DM a complement-dependent humoral process thought to be initiated by antibodies to endothelial cells results in a microangiopathy with secondary ischemic changes in muscles. On the other hand, in PM and IBM there is a T-cell response with invasion of muscle fibers by CD8+ lymphocytes and perforin-mediated cytotoxic necrosis. In IBM degenerative changes are also a feature and comprise autophagia with rimmed vacuole formation and inclusions containing β-amyloid and other proteins whose accumulation may be linked to impaired proteasomal function. The relationship between the inflammatory and degenerative component remains unclear, as does the basis for the selective vulnerability of certain muscles and the resistance to conventional forms of immunotherapy in most cases of IBM. Patients with DM or PM usually respond to treatment with glucocorticoids and immunosuppressive agents but their use remains largely empirical. Intravenous immunoglobulin therapy can be used to achieve disease control in patients with severe weakness or dysphagia, or in patients with immunodeficiency, but its use is limited by expense. Emerging therapies for resistant cases include TNFα inhibitors (etanercept, infliximab) and monoclonal antibodies (rituximab, alemtuzumab). However, experience with these therapies is still limited and there is a need for randomized trials to test their efficacy and establish guidelines for their use in clinical practice.
Keywords: Dermatomyositis, inclusion body myositis, inflammatory myopathies, pathogenesis, polymyositis, treatment
Introduction
The inflammatory myopathies are a heterogeneous group of diseases with diverse clinicopathological features and etiologies. The latest classification of these disorders is shown in Table 1. Focal or at times more widespread forms of myositis can be caused by viral, bacterial, fungal, protozoal or parasitic microorganisms and the clinical and pathological features and treatment of these infective forms of myositis are well-documented in other reviews. [1],[2] The present review will deal with those forms of myositis which have an immune basis, which are the ones most commonly encountered in neurological practice, [3] and will consider the latest pathogenetic concepts as well as current approaches to their diagnosis and treatment.
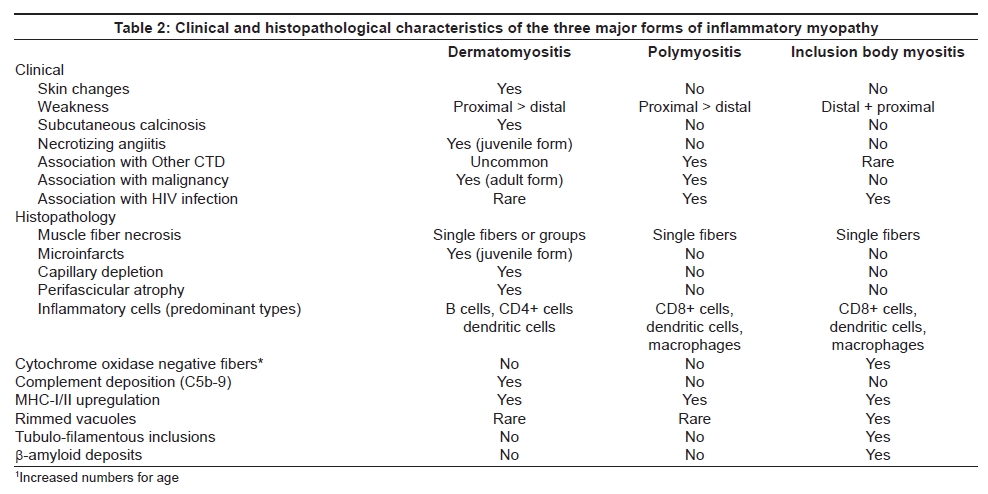
Classification As shown in Table 1 and Table 2, these conditions fall into a number of diagnostic categories on clinical and pathological grounds. The three major forms are: dermatomyositis (DM) and polymyositis (PM), both of which can occur in isolation or as part of a systemic connective tissue disease (′overlap syndrome′), and inclusion body myositis (IBM). The relative frequency of these conditions varies in different populations but it is generally recognized that DM and IBM are the most common forms, while PM is uncommon in isolation but is more likely to occur as part of a more widespread overlap syndrome. Sporadic IBM is now recognized as being the most common progressive myopathy manifesting over the age of 40 years, with a reported prevalence of 5-13 per million in Caucasian populations in Europe, North America and Australia.[4] However, there is little prevalence data from other parts of the world and this warrants further investigation. Inclusion body myositis can rarely also be familial [5],[6] and should then be differentiated from the hereditary forms of inclusion body myopathy, such as that caused by mutations in the GNE gene, in which myositis does not occur. [7]
Clinical Features
Although the skeletal muscles are involved diffusely in each of the three main forms of inflammatory myopathy, different patterns of muscle involvement are recognizable and can be helpful diagnostically. In DM and PM the muscle weakness usually develops subacutely and is diffuse and non-selective, but with a proximal emphasis, and is more severe in the upper limbs in some cases of DM. Muscle pain and tenderness are sometimes present in DM but are often absent. A more restricted form of myositis involving the scapular, cervical or lower paraspinal muscles may occur in scleroderma and such patients may present with the ′dropped head′ sign or camptocormia. Involvement of the facial and ocular muscles is uncommon in any of the inflammatory myopathies but bulbar and respiratory muscle involvement may occur in severe cases. Dysphagia is common particularly in patients with IBM. [4]
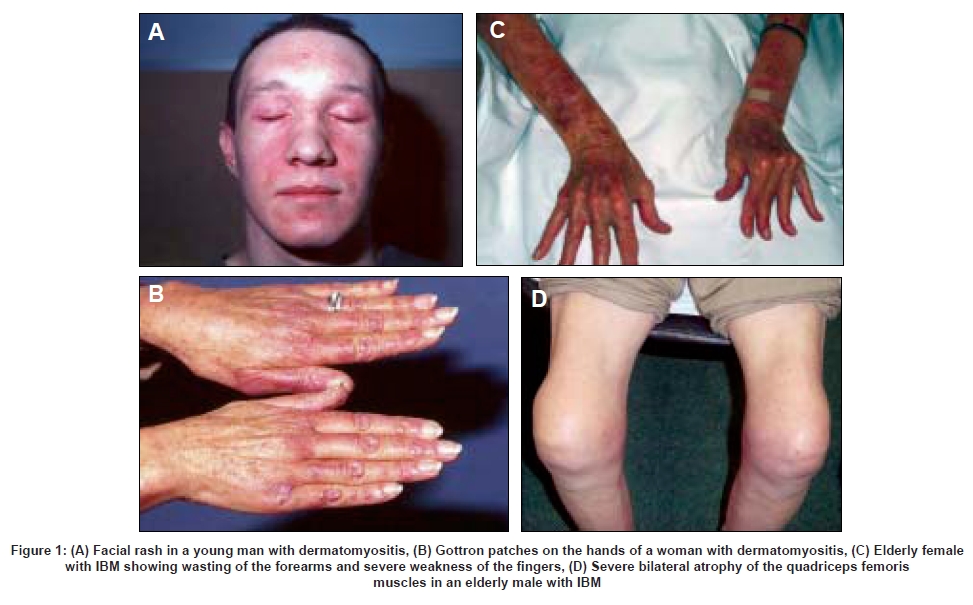
In contrast to the other forms of inflammatory myopathy which are more common in females, IBM more often affects males and has a slower and more insidious clinical course with a predilection for certain muscle groups. [4],[8] These include the quadriceps femoris and the forearm flexor muscles [Figure 1 and Figure 2] which undergo progressive weakness and atrophy during the course of the disease and are usually more severely affected on the non-dominant side of the body. [4],[9] The presenting symptoms include difficulty rising from chairs or walking up stairs, falls and difficulty in manipulating objects with the fingers. The order in which these muscle groups are affected is variable and, with progression of the disease, other muscle groups are also affected with increasing disability and impairment of mobility leading to the need to use a walking aid or wheelchair after 10-15 years.
When present, the cutaneous changes of DM on the face (heliotrope rash), hands and elbows (Gottron′s patches) and trunk (shawl sign) are distinctive and diagnostic of that condition [Figure 1]. However, they may be inconspicuous or even absent at the time of presentation and are easily overlooked, particularly in individuals of dark complexion. Conversely, in some cases of DM the cutaneous changes predominate and muscle involvement is minor or absent (′amyopathic′ DM). Subcutaneous calcinosis may also occur, particularly in cases of juvenile DM, but also in the adult form, and may be severe and widespread.
Association with Malignancy
The association with malignancies of various types is seen particularly in adult patients with DM. The risk of a malignancy is greatest in the five-year period preceding presentation and in the first five years after diagnosis of the myositis. [10],[11] It is therefore important that such patients should undergo a thorough malignancy screen at the time of presentation, and at least annually thereafter, or if a relapse occurs during the course of treatment of the myositis. This should include a careful clinical examination, including rectal and vaginal examination, mammography and pelvic ultrasound in females, and chest radiography. In addition, if there are any specific gastrointestinal, genitourinary or abdominal symptoms abdominal CT scans and endoscopic studies should be performed. Screening of the nasopharynx is also important, particularly in individuals of Asian origin who have a propensity to develop nasopharyngeal carcinoma. It is also important to screen for a malignancy in cases of ′necrotizing myopathy′, in which inflammatory changes in the muscle biopsy are absent or inconspicuous, as this form of myopathy is often paraneoplastic. [12]
There is no recognized association between IBM and malignancy, but up to 20% of such patients may have a paraproteinemia or an associated connective tissue disease or other autoimmune disease and the condition can also occur in individuals with HIV, HTLV-1 infection. [13],[14] Laboratory screening for a paraproteinemia, retroviral infection and these other conditions should therefore be carried out in all cases.
Diagnosis
Accurate diagnosis of the type of inflammatory myopathy is important as it will provide a guide to the response to treatment and prognosis in the individual patient. In addition, sets of diagnostic criteria for DM, PM and IBM based on a combination of clinical and pathological findings have been proposed for use in clinical trials and research studies. [15],[16]
An elevated serum creatine kinase (CK) level supports the diagnosis of an inflammatory myopathy but is nonspecific and the CK level may be normal or only mildly elevated in some cases of DM and in many cases of IBM. Electromyography is also helpful in reaching a diagnosis and may also provide an indication of the severity and extent of the myositis. The finding of myositis-specific antibodies (such as anti-Jo-1 or anti-signal recognition particle antibodies) are helpful as markers for forms of myositis that are often more resistant to treatment and have a poorer prognosis, but their diagnostic sensitivity is relatively low. [17] Anti-ribonuclear protein antibody is a marker for mixed connective tissue disease and anti-PM-Scl, which labels nucleoli, has a high specificity for the scleroderma-myositis overlap syndrome.
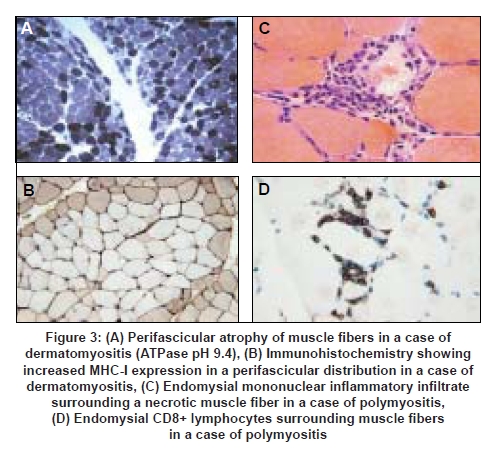
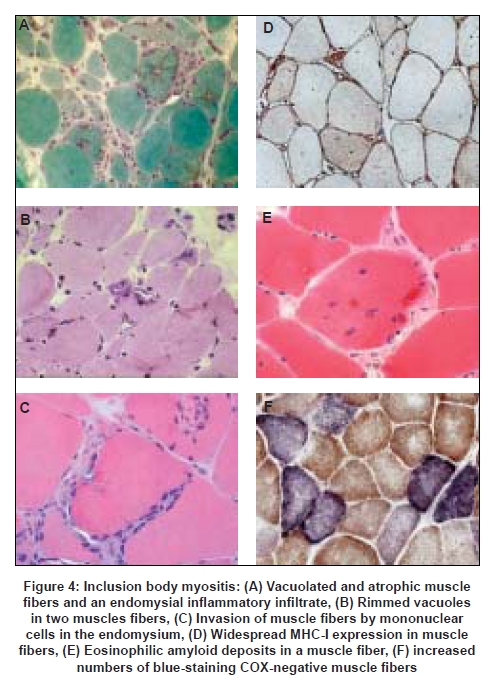
The definitive diagnostic procedure is the muscle biopsy which should ideally be performed prior to commencing treatment, and is usually taken from the vastus lateralis, deltoid or biceps brachii muscles, provided that the muscle selected is not too severely atrophied. For maximum information and diagnostic value the biopsy should be processed in a laboratory where the full range of histological and histochemical staining techniques are available, as well as immunohistochemical staining for complement (C5b-9) and MHC antigens. We recommend staining for both MHC-I and MHC-II as, in our experience, positive MHC-I staining alone is nonspecific. In addition, special stains for amyloid (Congo red viewed under Texas Red filters, or crystal violet) and phosphorylated tau (SMI-31 monoclonal antibody which recognizes tau as well as other neurofilament proteins) are helpful for confirming the diagnosis of IBM. The patterns of muscle fiber necrosis, inflammatory cell infiltrates and other histological changes differ in the three major types of inflammatory myopathies [see Table 2 and Figures 3 and 4] and careful examination of the biopsy will usually allow a definitive diagnosis to be reached in most cases provided that the tissue samples are of adequate size and the appropriate staining procedures are employed. The biopsy will also allow recognition of other less common varieties of myositis such as eosinophilic, granulomatous and vasculitic myositis.
Pathogenesis
It is now well established that DM, PM and IBM are distinct entities with different underlying pathogenetic mechanisms. This distinction is supported by the results of recent gene expression studies which have shown specific patterns of gene expression in different subtypes of inflammatory myopathy. [18],[19],[20],[21] A widely held model of DM implicates a complement-mediated attack on as yet unidentified endothelial cell antigens in muscle and skin as the cause of the distinctive form of microangiopathy and depletion of the muscle capillary bed which is a feature of this condition and muscle ischemia resulting in muscle fiber necrosis, microinfarcts and perifascicular atrophy. [22] A more recent model attributes greater importance to plasmacytoid dendritic cells and the induction of interferon α/β inducible proteins in endothelial cells and myofibres. [23] Deposition of the membranolytic C5b-9 complex in the walls of intramuscular arterioles and capillaries and increased expression of ICAM by capillary endothelial cells can be demonstrated immunohistochemically and muscle fibers also show increased expression of MHC-I and II antigens. The inflammatory infiltrate in DM is predominantly interstitial and perivascular and consists of B-cells, CD4+ T-cells as well as plasmacytoid dendritic cells [23] [Table 2]. In PM and IBM the inflammatory infiltrate is predominantly endomysial and consists of CD8+ and CD4+ cells as well as dendritic cells, macrophages and plasma cells. [18],[23],[24] There is invasion of non-necrotic muscle fibers expressing MHC-I antigens by CD8+ lymphocytes, [25] which induce cytotoxic necrosis through the liberation of perforins and granzymes. [26] The extent of the inflammatory infiltrate is variable and may be relatively inconspicuous in some cases of DM and IBM. As yet, the nature of the target antigens and the factors leading to the breakdown of immune tolerance in these conditions remain unknown. However, there is evidence that genetic factors, in particular the HLA-B08, DRB1FNx010301 and the 8.1 MHC ancestral haplotype play a part in conferring susceptibility to these diseases while other HLA alleles may be protective. [14]
In IBM the inflammatory changes tend to be more prominent early in the disease whereas the degenerative changes in muscle fibers (e.g. rimmed vacuoles, congophilic inclusions, tubulofilamentous aggregates) tend to be later manifestations and may be inconspicuous or even absent in biopsies from early cases. [27],[28] The significance of these degenerative changes, and their relationship to the inflammatory process in IBM, is still poorly understood. [29] Accumulation of β-amyloid in muscle fibers is suggested by some workers to play a central part in the pathogenesis of the disease [30] but is not specific to IBM, and a variety of other proteins such as phosphorylated tau and ubiquitin also accumulate in the muscle fiber inclusions, possibly as a result of protein misfolding and proteasomal dysfunction. In addition, potentially toxic mutant protein forms such as UBB +1 also accumulate as a result of aberrant transcription. [31] Another feature of IBM is the accumulation of multiple clonally-expanded somatic mtDNA mutations in segments of muscle fibers which lack cytochrome oxidase (COX) activity [32] [Figure 4]. Such fibers are present in greater numbers than in normal aging and could be one of the factors contributing to the muscular weakness and atrophy in IBM. [29] As in the case of other genetic and acquired myopathies there is as yet no satisfactory explanation for the differential vulnerability of different muscle groups in IBM. However, it has been proposed that this may be due to the existence of muscle-specific transcriptomes which determine the susceptibility of different muscle groups to the disease process. [29]
Treatment
The treatment of the inflammatory myopathies is still largely empirical as there is insufficient data to allow an evidence base. [3] Treatment is based on the use of glucocorticoids and other non-selective immunosuppressive and immunomodulatory therapies and, as yet, more specific forms of immunotherapy are not available. Few of the therapies have been subjected to randomized controlled trials (RCT) and few guidelines exist as to the optimal dose regimens, or the choice of agents to use in patients who fail to respond adequately to glucocorticoids.
Initial treatment
In the majority of cases of DM, PM and overlap syndromes, satisfactory disease control and an effective remission can be obtained with an initial course of prednisolone (~1 mg/Kg/day) in combination with an immunosuppressive agent from the start, or introduced within three to four weeks, which will allow earlier tapering of the steroid dose (e.g. by 5 mg/day/week to reach a dose of ~25-35 mg/day by the end of the second month, and then converting to an alternate-day regimen). Higher starting doses of prednisolone and slower rates of tapering are associated with a higher incidence of steroid side-effects and, in general, are best avoided. In patients who are severely affected, or if the diagnosis has been delayed, and in patients with weakness of the bulbar or respiratory muscles a more rapid response may be obtained by commencing treatment with intravenous methylprednisolone (0.5-1 g/day or every second day for three to six doses) followed by ongoing oral prednisolone. Either methotrexate (10-20 mg once per week) or azathioprine (2-3 mg/Kg/day) can be used for initial immunosuppression and appear to be equally effective. Patients who are less likely to respond well to treatment include the elderly, those with a long delay to diagnosis and commencement of treatment, and those with IBM or myositis associated with anti-synthetase or anti-SRP antibodies or malignancy.
Resistant cases
In patients who fail to respond to treatment it is important first to review the diagnosis and to exclude other conditions such as a metabolic, endocrine or toxic myopathy, which can sometimes mimic an inflammatory myopathy, or genetic disorders such as dystrophinopathy or muscular dystrophy (e.g. dystrophinopathy or facioscapulohumeral dystrophy) in which inflammatory changes are sometimes present in the biopsy. [33],[34] Having done so, other treatment options can then be considered [Figure 5]. In the first instance it is worth adding a second immunosuppressive agent (e.g. azathioprine if the patient is already on methotrexate, or vice versa) as the combination can be more effective than either agent alone. In addition, the possibility of a steroid myopathy should be considered and, if necessary, the dose of prednisolone should be reduced. If there is no improvement after six to eight weeks a further option is to substitute mycophenolate (0.5-1 g twice daily) for azathioprine. [35] If this is not effective more potent immunosuppressives such as the calcineurin inhibitors (cyclosporine 3-5 mg/kg/day or tacrolimus 0.1 mg/kg/day) which have a more selective action on T-cells, or pulse therapy with cyclophosphamide (0.5-1 g intravenously every two to four weeks), which has a more selective action on B-cells, may need to be considered, particularly in patients with the anti-synthetase syndrome who often have refractory myositis and interstitial lung disease, or those with anti-SRP antibodies who typically have a severe necrotizing myopathy with little inflammation and respond poorly to treatment.
Intravenous immunoglobulin therapy
Immunoglobulin therapy has been shown to be effective in resistant cases of DM in a RCT and also in uncontrolled trials in PM and overlap syndromes.[3] However, because of its limited availability and expense it is generally reserved for the treatment of severe cases, especially those with weakness of the bulbar or respiratory muscles who have failed to respond to glucocorticoids and immunosuppression or are intolerant to these forms of treatment. In patients with severe dysphagia which interferes with adequate nutrition IVIg therapy may lead to dramatic improvement in swallowing and may avoid the need for a percutaneous gastrostomy. Immunoglobulin therapy is also the treatment of choice in patients with myositis who are immunodeficient. [36]
The initial course of 2 g/Kg is usually administered over a five-day period (0.4 g/kg/day), followed by monthly three-day courses for a period of three to six months during which prednisolone and immunosuppressive agents are also continued. If there is no improvement after the first three courses of IVIg therapy it should be discontinued.
Emerging therapies
There have been a number of reports of successful treatment of refractory cases of DM and PM with the TNF-α antagonists infliximab and etanercept which are not yet widely available (see review by Baer 2006). [35] The response in these cases has been variable and their effectiveness needs to be confirmed in a RCT. The B-cell depleting monoclonal antibody rituximab has also been shown to be effective in some DM patients with refractory disease, in keeping with the concept that the condition is due to a B-cell-mediated humoral immune process. [37] It has also been found to be effective in some cases of refractory PM, including those with anti-synthetase antibodies. [35] However, experience with these agents is still limited and they need to be evaluated further in randomized controlled trials. Other monoclonal antibodies against the C5 component of complement (eculizumab) and against T-cells (alemtuzumab) also have a potential application in resistant cases of DM and PM and warrant further evaluation. [38] Autologous hematopoietic stem cell transplantation has been reported to be effective in a case of severe anti-Jo-1 associated myositis and interstitial lung disease and is the last resort for severely disabled patients who are unresponsive to all other forms of treatment. [39]
Inclusion body myositis
Inclusion body myositis is the least responsive of the inflammatory myopathies to conventional immunotherapy and there is an urgent need to find more effective forms of treatment to prevent the inexorable progression of the disease. [14] A minority of cases do respond to treatment with glucocorticoids and immunosuppression, particularly those with an associated autoimmune disease such as Sjogren′s syndrome, but the response is usually only temporary. Nevertheless, a three to six month trial of prednisolone (0.5-1 mg/kg/day or on alternate days) in combination with methotrexate, azathioprine or mycophenolate can be offered provided that the patient′s general medical condition is satisfactory and that the patient is monitored regularly to detect any adverse effects such as steroid-induced aggravation of the muscle weakness. Other immunosuppressive agents have not been adequately evaluated but are usually ineffective in practice. Intravenous immunoglobulin therapy can improve swallowing in patients with severe dysphagia [40] and has been found to prevent disease progression in a six-month RCT in Germany, [41] but the long-term benefits of such treatment have yet to be investigated. Improvement or slowing of disease progression has been reported in small trials of lymphocyte depletion using anti-T lymphocyte globulin [42] or alemtuzumab, [43] and of the TNF-a antagonist etanercept, [44] but the effectiveness of these therapies and their side-effect profile require further evaluation.
References
| 1. | Mastaglia FL. Inflammatory diseases of muscle. Oxford: Blackwell Scientific Publications; 1988. Back to cited text no. 1 |
| 2. | Chimelli L. Infective myopathies. In: Mastaglia FL, Hilton-Jones D, editors. Handbook of Clinical Neurology: Myopathies. Amsterdam: Elsevier; 2007. p. 303-19. Back to cited text no. 2 |
| 3. | Mastaglia FL, Garlepp MJ, Phillips BA, Zilko PJ. Inflammatory myopathies: Clinical, diagnostic and therapeutic aspects. Muscle Nerve 2003;27:407-25. Back to cited text no. 3 [PUBMED] [FULLTEXT] |
| 4. | Needham M, James I, Corbett A, Day T, Christiansen F, Phillips B, et al. Sporadic inclusion body myositis: Phenotypic variability and influence of HLA-DR3 in a cohort of 57 Australian cases. J Neurol Neurosurg Psychiatry 2008;79:1056-60. Back to cited text no. 4 [PUBMED] [FULLTEXT] |
| 5. | Mastaglia F, Price P, Walters S, Fabian V, Miller J, Zilko P. Familial inclusion body myositis in a mother and son with different ancestral MHC haplotypes. Neuromuscul Disord 2006;16:754-8. Back to cited text no. 5 [PUBMED] [FULLTEXT] |
| 6. | Sivakumar K, Semino-Mora C, Dalakas MC. An inflammatory, familial, inclusion body myositis with autoimmune features and a phenotype identical to sporadic inclusion body myositis: Studies in three families. Brain 1997;120:653-61. Back to cited text no. 6 [PUBMED] [FULLTEXT] |
| 7. | Argov Z, Mitrani-Rosenbaum S. Hereditary inclusion body myopathy and other rimmed vacuolar myopathies. In: Mastaglia FL, Hilton-Jones D, editors. Handbook of clinical neurology. Vol. 86. Amsterdam: Elsevier B.V.; 2007. p. 243-53. Back to cited text no. 7 |
| 8. | Badrising UA, Maat-Schieman ML, van Houwelingen JC, van Doorn PA, van Duinen SG, van Engelen BG, et al. Inclusion body myositis: Clinical features and clinical course of the disease in 64 patients. J Neurol 2005;252:1448-54. Back to cited text no. 8 [PUBMED] [FULLTEXT] |
| 9. | Phillips BA, Cala LA, Thickbroom GW, Melsom A, Zilko PJ, Mastaglia FL. Patterns of muscle involvement in inclusion body myositis: Clinical and magnetic resonance imaging study. Muscle Nerve 2001;24:1526-34. Back to cited text no. 9 [PUBMED] [FULLTEXT] |
| 10. | Yazici Y, Kagen LJ. The association of malignancy with myositis. Curr Opin Rheumatol 2000;12:498-500. Back to cited text no. 10 [PUBMED] [FULLTEXT] |
| 11. | Zantos D, Zhang Y, Felson D. The overall and temporal association of cancer with polymyositis and dermatomyositis. J Rheumatol 1994;21:1855-9. Back to cited text no. 11 [PUBMED] |
| 12. | Levin MI, Mozaffar T, Al-Lozi MT, Pestronk A. Paraneoplastic necrotizing myopathy: Clinical and pathological features. Neurology 1998;50:764-7. Back to cited text no. 12 [PUBMED] |
| 13. | Cupler EJ, Leon-Monzon M, Miller J, Semino-Mora C, Anderson TL, Dalakas MC. Inclusion body myositis in HIV-1 and HTLV-1 infected patients. Brain 1996;119:1887-93. Back to cited text no. 13 [PUBMED] [FULLTEXT] |
| 14. | Needham M, Mastaglia FL. Inclusion body myositis: Current pathogenetic concepts and diagnostic and therapeutic approaches. Lancet Neurol 2007;6:620-31. Back to cited text no. 14 [PUBMED] [FULLTEXT] |
| 15. | Mastaglia FL, Phillips BA. Idiopathic inflammatory myopathies: Epidemiology, classification, and diagnostic criteria. Rheum Dis Clin North Am 2002;28:723-41. Back to cited text no. 15 [PUBMED] |
| 16. | Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, et al. 119th ENMC international workshop: Trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10-12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004;14:337-45. Back to cited text no. 16 [PUBMED] [FULLTEXT] |
| 17. | Garlepp MJ, Mastaglia FL. Autoantibodies in inflammatory myopathies. Am J Med Sci 2000;319:227-33. Back to cited text no. 17 [PUBMED] [FULLTEXT] |
| 18. | Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, et al. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol 2005;57:664-78. Back to cited text no. 18 [PUBMED] [FULLTEXT] |
| 19. | Greenberg SA, Sanoudou D, Haslett JN, Kohane IS, Kunkel LM, Beggs AH, et al. Molecular profiles of inflammatory myopathies. Neurology 2002;59:1170-82. Back to cited text no. 19 [PUBMED] [FULLTEXT] |
| 20. | Raju R, Dalakas MC. Gene expression profile in the muscles of patients with inflammatory myopathies: Effect of therapy with IVIg and biological validation of clinically relevant genes. Brain 2005;128:1887-96. Back to cited text no. 20 [PUBMED] [FULLTEXT] |
| 21. | Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan D, Bremer EG, et al. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: A novel model of pathogenesis. J Immunol 2002;168:4154-63. Back to cited text no. 21 [PUBMED] [FULLTEXT] |
| 22. | Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003;362:971-82. Back to cited text no. 22 [PUBMED] [FULLTEXT] |
| 23. | Greenberg SA. Proposed immunologic models of the inflammatory myopathies and potential therapeutic implications. Neurology 2007;69:2008-19. Back to cited text no. 23 [PUBMED] [FULLTEXT] |
| 24. | Greenberg SA, Bradshaw EM, Pinkus JL, Pinkus GS, Burleson T, Due B, et al. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology 2005;65:1782-7. Back to cited text no. 24 [PUBMED] [FULLTEXT] |
| 25. | Engel AG, Arahata K. Monoclonal antibody analysis of mononuclear cells in myopathies, II: Phenotypes of autoinvasive cells in polymyositis and inclusion body myositis. Ann Neurol 1984;16:209-15. Back to cited text no. 25 [PUBMED] |
| 26. | Goebels N, Michaelis D, Engelhardt M, Huber S, Bender A, Pongratz D, et al. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest 1996;97:2905-10. Back to cited text no. 26 [PUBMED] [FULLTEXT] |
| 27. | Blume G, Pestronk A, Frank B, Johns DR. Polymyositis with cytochrome oxidase negative muscle fibres: Early quadriceps weakness and poor response to immunosuppressive therapy. Brain 1997;120:39-45. Back to cited text no. 27 [PUBMED] [FULLTEXT] |
| 28. | Chahin N, Engel AG. Correlation of muscle biopsy, clinical course, and outcome in PM and sporadic IBM. Neurology 2008;70:418-24. Back to cited text no. 28 [PUBMED] [FULLTEXT] |
| 29. | Needham M, Mastaglia FL. Sporadic inclusion body myositis: A continuing puzzle. Neuromuscul Disord 2008;18:6-16. Back to cited text no. 29 [PUBMED] [FULLTEXT] |
| 30. | Askanas V, Engel WK. Inclusion-body myositis: A myodegenerative conformational disorder associated with Abeta, protein misfolding, and proteasome inhibition. Neurology 2006;66:S39-48. Back to cited text no. 30 [PUBMED] [FULLTEXT] |
| 31. | Fratta P, Engel WK, Van Leeuwen FW, Hol EM, Vattemi G, Askanas V. Mutant ubiquitin UBB+1 is accumulated in sporadic inclusion-body myositis muscle fibers. Neurology 2004;63:1114-7. Back to cited text no. 31 [PUBMED] [FULLTEXT] |
| 32. | Oldfors A, Moslemi AR, Jonasson L, Ohlsson M, Kollberg G, Lindberg C. Mitochondrial abnormalities in inclusion-body myositis. Neurology 2006;66:S49-55. Back to cited text no. 32 [PUBMED] [FULLTEXT] |
| 33. | Mastaglia FL. When the treatment does not work: Polymyositis. Pract Neurol 2008;In press. Back to cited text no. 33 |
| 34. | Nirmalananthan N, Holton JL, Hanna MG. Is it really myositis? A consideration of the differential diagnosis. Curr Opin Rheumatol 2004;16:684-91. Back to cited text no. 34 |
| 35. | Baer AN. Advances in the therapy of idiopathic inflammatory myopathies. Curr Opin Rheumatol 2006;18:236-41. Back to cited text no. 35 [PUBMED] [FULLTEXT] |
| 36. | Dalakas MC. Retroviruses and inflammatory myopathies in humans and primates. Baillieres Clin Neurol 1993;2:659-91. Back to cited text no. 36 [PUBMED] |
| 37. | Levine TD. Rituximab in the treatment of dermatomyositis: An open-label pilot study. Arthritis Rheum 2005;52:601-7. Back to cited text no. 37 [PUBMED] [FULLTEXT] |
| 38. | Dalakas MC. Therapeutic targets in patients with inflammatory myopathies: Present approaches and a look to the future. Neuromuscul Disord 2006;16:223-36. Back to cited text no. 38 [PUBMED] [FULLTEXT] |
| 39. | Baron F, Ribbens C, Kaye O, Fillet G, Malaise M, Beguin Y. Effective treatment of Jo-1-associated polymyositis with T-cell-depleted autologous peripheral blood stem cell transplantation. Br J Haematol 2000;110:339-42. Back to cited text no. 39 [PUBMED] [FULLTEXT] |
| 40. | Cherin P, Pelletier S, Teixeira A, Laforet P, Simon A, Herson S, et al. Intravenous immunoglobulin for dysphagia of inclusion body myositis. Neurology 2002;58:326. Back to cited text no. 40 [PUBMED] [FULLTEXT] |
| 41. | Walter MC, Lochmuller H, Toepfer M, Schlotter B, Reilich P, Schroder M, et al. High-dose immunoglobulin therapy in sporadic inclusion body myositis: A double-blind, placebo-controlled study. J Neurol 2000;247:22-8. Back to cited text no. 41 |
| 42. | Lindberg C, Trysberg E, Tarkowski A, Oldfors A. Anti-T-lymphocyte globulin treatment in inclusion body myositis: A randomized pilot study. Neurology 2003;61:260-2. Back to cited text no. 42 [PUBMED] [FULLTEXT] |
| 43. | Dalakas MC, Rakocevic G, McElroy B, Saljegheh M, Brookline MA, Love MH, et al. Alemtuzumab (CAMPATH 1-h) therapy in sporadic inclusion body myositis (sIBM): A treatment trial in patients with established natural history data. Neurology 2007;68:A361. Back to cited text no. 43 |
| 44. | Barohn RJ, Herbelin L, Kissel JT, King W, McVey AL, Saperstein DS, et al. Pilot trial of etanercept in the treatment of inclusion-body myositis. Neurology 2006;66:S123-4. Back to cited text no. 44 [PUBMED] [FULLTEXT] |
Copyright 2008 - Neurology India
The following images related to this document are available:
Photo images
[ni08076t2.jpg]
[ni08076f2.jpg]
[ni08076f4.jpg]
[ni08076t1.jpg]
[ni08076f5.jpg]
[ni08076f3.jpg]
[ni08076f1.jpg]
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}