
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 56, Num. 3, 2008, pp. 271-280
|
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 271-280
Review Article
How to go about diagnosing and managing the limb-girdle muscular dystrophies
Guglieri Michela, Bushby Kate
Department of Genetics, Institute of Human Genetics, International Centre for Life, Central Parkway, Newcastle upon Tyne NE1 3BZ
Correspondence Address:Institute of Human Genetics, International Centre for Life, Central Parkway, Newcastle upon Tyne NE1 3BZ
kate.bushby@ncl.ac.uk
Date of Acceptance: 24-Jul-2008
Code Number: ni08077
Abstract
The increasing knowledge about limb girdle muscular dystrophy (LGMD) has clarified many aspects of this extensive group of neuromuscular conditions and has moreover proven their wide clinical and genetic heterogeneity. For these reasons, achieving a precise diagnosis of a particular type of LGMD may be still difficult and requires a comprehensive approach, which includes epidemiology, medical history, clinical examination, laboratory and genetic tests. All of the LGMDs are individually rare and their population frequency is highly variable. The prevalence of the different forms of LGMD in different populations has to be considered for the differential diagnosis. Some characteristic clinical features may help to distinguish subtypes of LGMD however protein analysis on muscle biopsy and genetic testing still represent the gold standard in the diagnosis of these muscular dystrophies. Reaching a precise diagnosis in all patients is important to allow genetic counseling to be properly applied and to direct appropriate medical management with a potential positive impact on the length and quality of life. Moreover, new specific therapeutic approaches, including limited local gene therapy, have been emerging over the last few years and require a precise genetic definition of the conditions. This article will concentrate on the diagnostic process by which these disorders can be defined and the implications of making these diagnoses.
Keywords: Diagnosis, limb girdle muscular dystrophies
Introduction
Trying to reach a precise diagnosis in a patient with a suspected limb girdle muscular dystrophy (LGMD) is traditionally regarded as difficult. Although the increasing knowledge about the genetic basis of the different diseases in this group has made the group seem rather complicated, it also allows us the possibility of reaching a proper diagnosis. The diagnostic process needs to take into account the clinical history and examination of the patient, as well as a combination of possible specialized analysis of the muscle biopsy and DNA.
A possible diagnosis of LGMD should be considered in a patient with muscular dystrophy having predominant involvement of the "limb-girdle" (pelvic and shoulder) musculature. However, these clinical findings are not unique to the LGMDs and other neuromuscular disorders such as Becker muscular dystrophy, late onset spinal muscular atrophy, myotonic dystrophy Type 2, Bethlem myopathy and Pompe′s disease may all show overlapping clinical features and rarer disorders such as congenital myasthenic syndromes may also need to be considered in the differential diagnosis. On the other hand, for many of the genes involved in LGMDs, a broad heterogeneity of clinical presentation and muscle involvement has emerged. This is particularly true for some of the dominant disorders in the LGMD classification, such as LGMD1A where myotilin mutations are associated also with the disorders known as the myofibrillar myopathies, [1],[2] LGMD1B where lamin A/C mutations can also cause a wide range of diseases [3],[4] and LGMD1C where the clinical presentation includes hyperCKaemia, rippling muscle disease, myalgia and distal myopathy. [5],[6] This means that there needs to be a high level of suspicion in this group of disorders if the correct diagnosis is going to be reached.
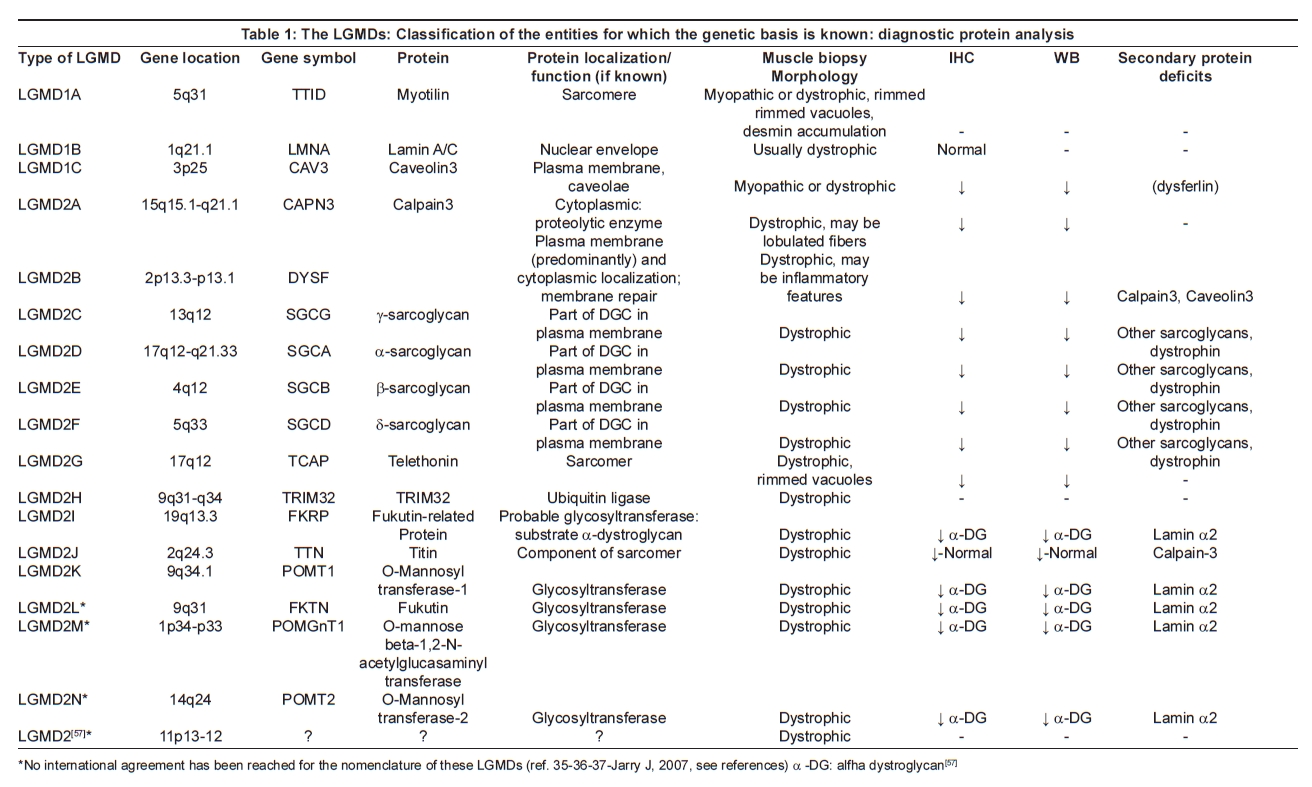
The molecular basis of the diseases is also highly heterogeneous. The process of gene identification in LGMDs has involved a combination of linkage and candidate gene analysis, resulting in a gene and protein-based classification which includes three known genes causing autosomal dominant LGMDs [7] and 14 known genes causing autosomal recessive LGMDs [8],[9] [Table 1].
The genes and proteins causing the diseases in these groups show a huge range of localization across the muscle fiber, from sarcolemma to nuclear envelope, with functions ranging from structural to enzymatic. Despite several comprehensive studies developed over the last few years, the genetic etiology of many cases of LGMD is not yet known.
Considering the wide clinical and genetic variability of LGMDs, achieving a precise diagnosis, especially in sporadic patients, may be difficult and requires a comprehensive clinical and laboratory approach. The definition of a precise diagnosis is increasingly allowing directed management for these diseases by the ability to predict specific complications such as those of the cardiac or respiratory systems. [10]
Proper genetic counseling can only be offered if the precise mode of inheritance is known which often depends on a genetic diagnosis and prenatal diagnosis can only be provided if the underlying molecular defect for the condition is known. Finally, in the future a precise genetic diagnosis will be the starting point for specific gene and protein-based therapies.
This article will concentrate on the diagnostic process by which these disorders can be defined and the implications of making these diagnoses.
History and Examination
Limb girdle muscular dystrophy (LGMD) was first designated as a disease group to address a set of patients with predominantly proximal muscle weakness and a dystrophic pattern on muscle biopsy. It separated them from the more classically recognized "Duchenne/ Becker" and facioscapulohumeral muscular dystrophies, both of which are much more common than LGMDs in most populations, and for which relatively straightforward diagnostic tests now exist. The key hallmarks of LGMD remain the marked weakness of the pelvic and shoulder girdle muscles; however, limb girdle muscle weakness is a primary problem in a large number of patients with very variable underlying muscular conditions. Moreover, the clinical course of LGMD ranges from severe forms with early onset and rapid progression to milder forms with late onset.
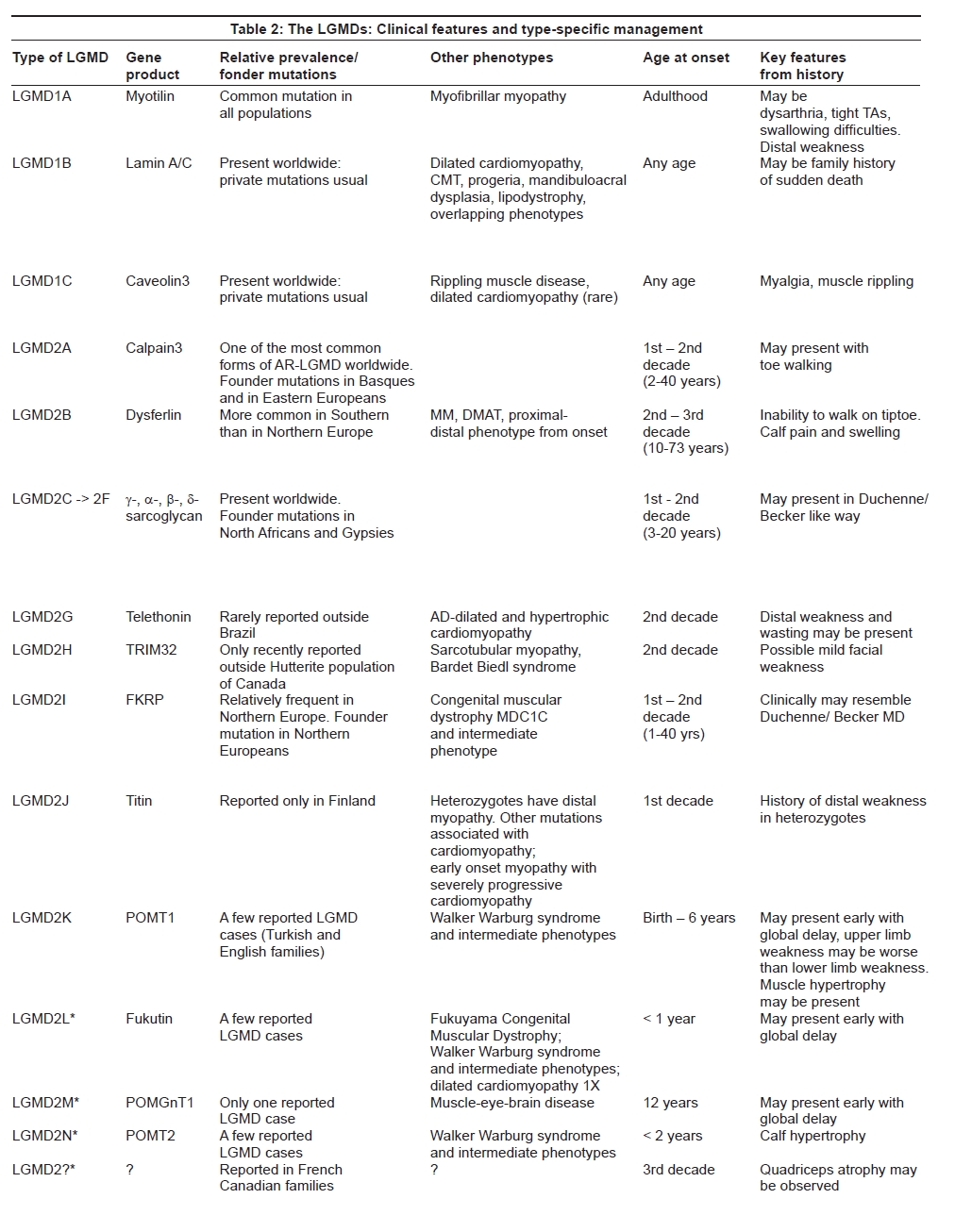
As the different disease entities within the group have become better defined based on their distinct genetic basis, accompanying clinical features may help to distinguish subtypes of LGMD. [11] Particular features which need to be paid attention to include muscle hypertrophy or atrophy, scapular winging, muscle rippling and contractures. While the history will in many cases be a relatively nonspecific story of progressive proximal weakness, with onset at any age, there are some clues in the history of onset which can be very suggestive of a particular diagnosis. Onset, distribution of muscle weakness and wasting and other characteristic clinical features can also help in the differential diagnosis of the different forms of LGMD [Table 2a and b].
Autosomal dominant limb girdle muscular dystrophy (LGMD1)
Most of the autosomal dominant LGMDs have been described in single extended pedigrees and are considered very rare. Moreover, among the genetic causes of the autosomal dominant LGMDs, pure limb-girdle weakness appears to be a rare phenotype while presentations with a distal myopathy or myofibrillar myopathy are increasingly recognized. The variability in presentation for all of the LGMDs means that different family members, or indeed the same individual, may present with one or more manifestations of mutation in a particular gene. Consequently, in some of the autosomal dominant types of LGMD the family history may be particularly informative for the diagnosis. For example, the presence of respiratory failure, dysarthria or cardiomyopathy in the index case or wider family may help to suggest LGMD1A. [12] History of arrhythmia or contractures can be suggestive of laminopathy or LGMD1B, [13] while a history of muscle rippling or myalgia may be suggestive of LGMD1C [14] [Table 2a and b].
LGMD2A
The clinical examination and a careful observation of the pattern of muscle weakness may be very helpful in recognizing this particular type of LGMD. In particular, a striking and early involvement of the posterior thigh muscles (adductors, semimembranosus and vastus intermedius muscles) with relative sparing of the vastus lateralis, sartorius and gracilis [15] appears to be almost stereotypic of calpainopathy. The presence of early contractures, scapular winging and preserved respiratory function is in contradiction to most other types of LGMD and can be very helpful in pinpointing the diagnosis. [16] Patients typically lose independent ambulation between 11 and 28 years after onset of the condition however disease progression in LGMD2A can be widely variable. [17]
LGMD2B
The clinical phenotype of dysferlinopathies is widely variable ranging from typical LGMD2B to Miyoshi Myopathy (MM) and distal anterior compartment myopathy (DMAT). [18] Over the last year other clinical presentations associated with mutations in the DYSF gene have been described, including an intermediate form with proximal and distal weakness from onset and patients with rigid spine and leg muscle stiffness with onset in the sixth decade. [19],[20],[21] Involvement of the shoulder girdle is usually a later event, although local biceps atrophy may be present at an early stage of the disease. Around 10% of LGMD2B patients experiences muscle pain and swelling, especially in the calves.
A history of normal childhood activities or even outstanding sporting ability until a relatively sudden onset in the late teens of muscle difficulties, often with early inability to stand on tiptoe, and sometimes with sudden onset of calf pain and swelling seems to be almost pathognomonic for the diagnosis. [22] This group of patients in particular may be misdiagnosed as myositis, which appears to be non-responsive to steroids.
LGMD2C->F (Sarcoglycanopathies)
Before the molecular analysis, sarcoglycanopathies were labeled "severe childhood autosomal recessive muscular dystrophy" (SCARMD) to describe children who showed a Duchenne-like clinical phenotype with an autosomal recessive mode of inheritance. [23] Similar to Duchenne and Becker muscular dystrophy, patients affected by sarcoglycanopathies frequently show calf and tongue hypertrophy. However, an important distinguishing factor from dystrophinopathies is the absence of any cognitive impairment, in all types of sarcoglycanopathies.
Alpha-sarcoglycanopathy (LGMD2D) tends to be clinically milder compared to the other sarcoglycanopathies [24],[25] although no pathognomonic clinical features can distinguish the different sarcoglycanopathies.
LGMD2G
LGMD2G or Telethoninopathy is a rare form of AR-LGMD described only in a few Brazilian families. Clinically it appears to be characterized by mildly progressive proximal weakness in the upper and lower limbs and distal muscle involvement with foot drop. Calf hypertrophy is often present and can be asymmetric. [26]
LGMD2H
LGMD2H results from mutations in the TRIM32 gene and has been only recently described in non-Hutterite populations. [27],[28],[29]
Alpha-dystroglycanopathies
Defects in the glycosylation of alpha-dystroglycan (alpha-DG) are classically associated with congenital muscular dystrophies (CMDs). However, over the last few years new LGMD phenotypes associated with mutations in the six known glycosyltransferase genes have been emerging. [30] In this context, the most common form of LGMD due to abnormal glycosylation of alpha-DG is LGMD2I. It results from mutations in the Fukutin-related protein gene (FKRP). [31],[32] LGMD2I shows a high variability in clinical presentations, ranging from the severe Duchenne-like disease course to late-onset and slow progressive forms. Characteristic clinical features of this LGMD include calf and tongue hypertrophy. A response to steroid treatment (prednisolone) has been described in some patients with LGMD2I [33] and future clinical trials may clarify this observation.
Other LGMD phenotypes have been recently shown to be associated with mutations in other known or putative glycosyltransferase genes, in particular POMT1, POMT2, fukutin and POMGnT1. [34],[35],[36],[37] The observation of such intermediate phenotypes between LGMDs and congenital muscular dystrophies, the structural or functional brain involvement in some of these new forms of LGMD and the pathogenic background complicate the classification of these new phenotypes and genetic entities. [38]
LGMD2J
LGMD2J is a severe form of autosomal recessive muscular dystrophy which so far has been described only in the Finnish population. It is characterized by onset in the first to third decade with progressive weakness of all proximal muscles. Facial muscles are not involved but some patients develop late distal muscle weakness. [39]
Associated complication
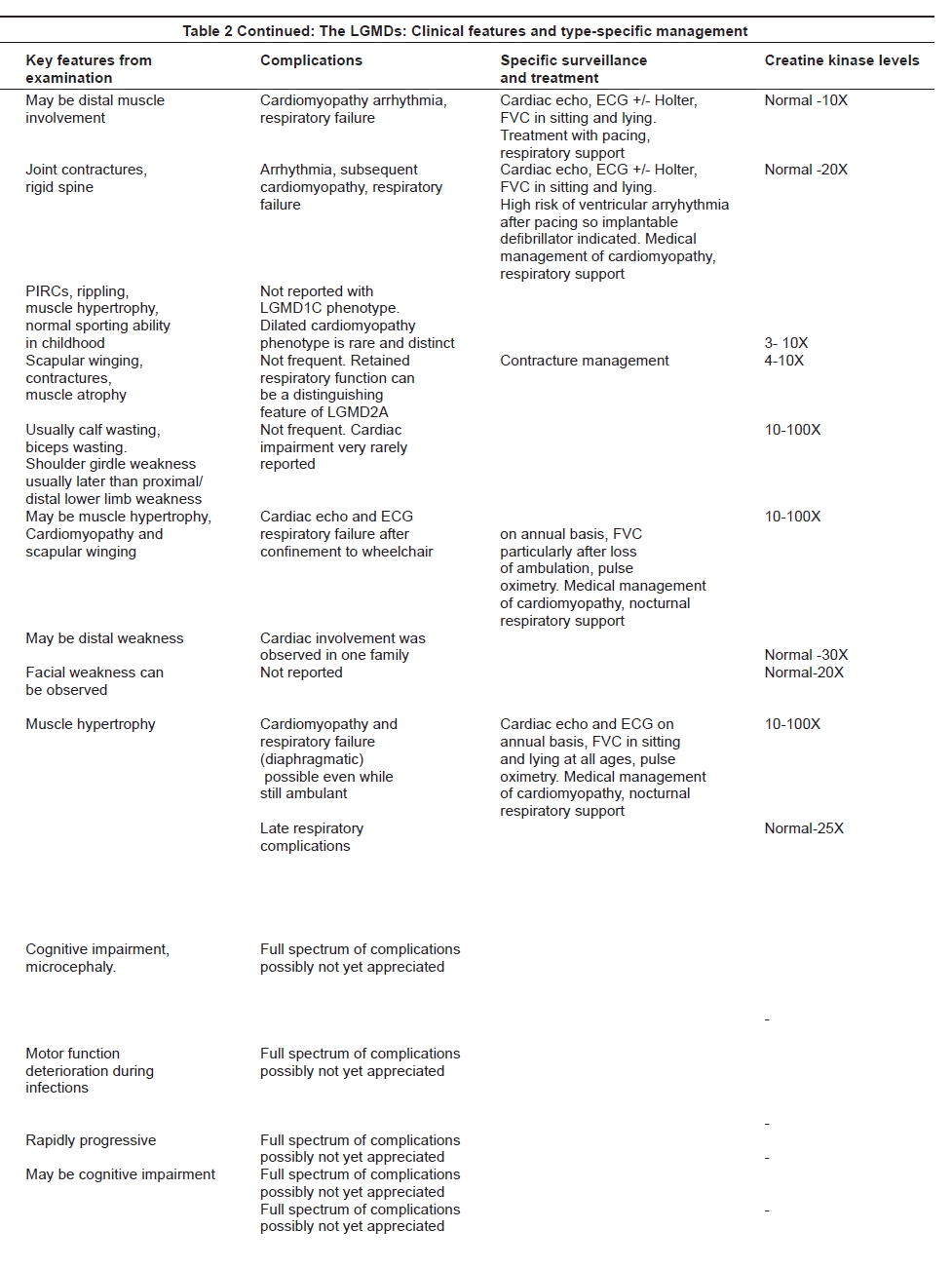
Muscular dystrophies are often multisystem disorders. Identification of associated complications can be helpful in the differential diagnosis of the different LGMD subtypes. Also, the recognition of a particular type of muscular dystrophy can help to assign a more precise risk of complications and to direct appropriate management and type-specific follow-up. The more frequent and severe complications in LGMDs include respiratory and heart involvement. The cardiovascular complications include the development of a progressive cardiomyopathy or arrhythmias, while respiratory complications may or may not have significant diaphragmatic weakness as a clear component of the problem. These complications are present at different frequencies within the various disorders in the LGMD group [Table 2a and b]. Preserved respiratory and heart function is a distinguishing clinical feature of LGMD2A. [16] Restrictive respiratory abnormalities and cardiomyopathy with reduced ejection fraction are almost constantly associated with LGMD2I and sarcoglycanopathies, with the exception of LGMD2D which is rarely complicated by heart involvement. [10] These complications need to be sought specifically and an appropriate management of any detected problems may have a positive impact on the length and quality of life. [40],[41]
The diagnostic process [Figure 1]
The first step to diagnosing a particular type of LGMD is knowledge of the prior probability of seeing any one of the particular subtypes in a particular population . All of the LGMDs are individually rare and some of them have only been described in a few families. Moreover, their population frequency is highly variable and the prevalence of the different forms of LGMD differs between populations, depending on geographical and ethnic factors. There are some problems in comparing the prevalence of the different types of LGMD as the ways used to define the different entities (for example, by protein diagnosis or by a combination of protein and DNA studies) as well as the denominators for the different studies are not uniform.
In the Caucasian population the autosomal dominant forms of LGMD seem to be responsible for around 14% of all cases but they are probably less common in other ethnicities. [11] LGMD2A seems to be the most frequent form of LGMD worldwide, though the relative frequency of the different autosomal recessive forms in different populations is still under investigation, and comparison from country to country is complicated by the fact that different studies report their findings in different ways.
In Brazil the relative proportion of the different forms among classified patients with AR-LGMD (through DNA and/or muscle protein analysis) from 120 families was found to be 32% for LGMD2A, 22% for LGMD2B, 32% for the sarcoglycanopathy group, 3% for LGMD2G and 11% for LGMD2I. [42] In the Italian population, looking at both dominant and recessive diagnoses, LGMD2A was the most common with 28.4%, dysferlinopathy 18.7%, sarcoglycanopathy 18.1%, LGMD2I 6.4%, LGMD1C 1.3% and undetermined diagnosis 27.1%.[43] In the North of England population, LGMD2A represented 26.5% of the whole LGMD group, LGMD2I 19.1%, sarcoglycanopathy 11.7%, LGMD2B 5.9% and unclassified LGMD constituted 27.9% (Norwood et al. , in preparation). In this population, LGMDs as a whole represented 6.15% of the total clinic population (when the most common diagnoses were myotonic dystrophy and dystrophinopathies). In Australia, calpainopathy represented 8% of a total muscular dystrophy population and dysferlinopathy 5%, while LGMD2I was less frequent (3%). [44] By contrast, LGMD2I represents a common type of LGMD in the German and Scandinavian populations. [45] The most common LGMDs in the United States are calpainopathies, dysferlinopathies, sarcoglycanopathies and dystroglycanopathies [46] with a distribution of immunophenotypes of 12% calpainopathy, 18% dysferlinopathy, 15% sarcoglycanopathy, 15% dystroglycanopathy and 1.5% caveolinopathy. In the Netherlands, LGMD2A is the most frequent type of LGMD, consisting of 21% of the classified families. LGMD2B is rare, while sarcoglycanopathies account for 16% of the classified families and of 69% of these have a severe, Duchenne like, autosomal recessive LGMD phenotype. LGMD2I consists of 8% of the genetically defined cases in this population. [47] Data from other populations are scanty given the lack in many areas of access to specialized diagnostic facilities.
None of the different types of LGMD can be distinguished on basic muscle histology or electrophysiology. Electromyography may be useful in distinguishing this myopathic group of diseases from myotonic dystrophy Type2 (PROMM, DM2) which may be clinically similar. Morphological pattern of the muscle biopsy may be crucial in distinguishing LGMDs from Pompe′s disease or other metabolic syndromes.
The serum creatine kinase (CK) is variably elevated in the different disorders and this may serve as a useful indicator to subdivide the different types of LGMD, though it is no more than a guide. As a rule of thumb, the autosomal recessive types of LGMD are typically associated with a much higher level of CK elevation than the dominant forms of LGMD, with the exception of LGMD1C. Among the recessive forms, LGMD2B, 2I and sarcoglycanopathies are usually associated with very high CK levels.
Over the last few years muscle magnetic resonance imaging (MRI) has been increasingly applied in order to determine distinct patterns of muscle involvement and may represent a promising advance in the differential diagnosis of neuromuscular conditions and of the different forms of LGMD and in directing appropriate protein and genetic investigations. [15],[48],[49]
Despite the new knowledge about the molecular basis of most of the LGMDs, a diagnostic approach exclusively based on genetic testing is not usually applicable as it is often inefficient and expensive. The muscle biopsy is unavoidable in most cases and still represents an informative and economic step in the diagnostic process. Diagnosis therefore relies on a combination of clinical assessment, specialized muscle immunoanalysis and genetic testing. In informative families, linkage analysis may be useful to identify the gene involved. Otherwise, protein analysis can be used to pinpoint the likely gene primarily involved, though often complex patterns of secondary protein involvement in addition to the primary defect can complicate this process. A wide range of antibodies needs to be applied and frequently immunolabeling of a muscle biopsy sample needs to be supplemented by immunoblotting in order to elucidate primary and secondary protein abnormalities. No studies compare open versus needle biopsies, although it is important to obtain sufficient tissue to allow a comprehensive analysis and meaningful interpretation [10] [Table 1].
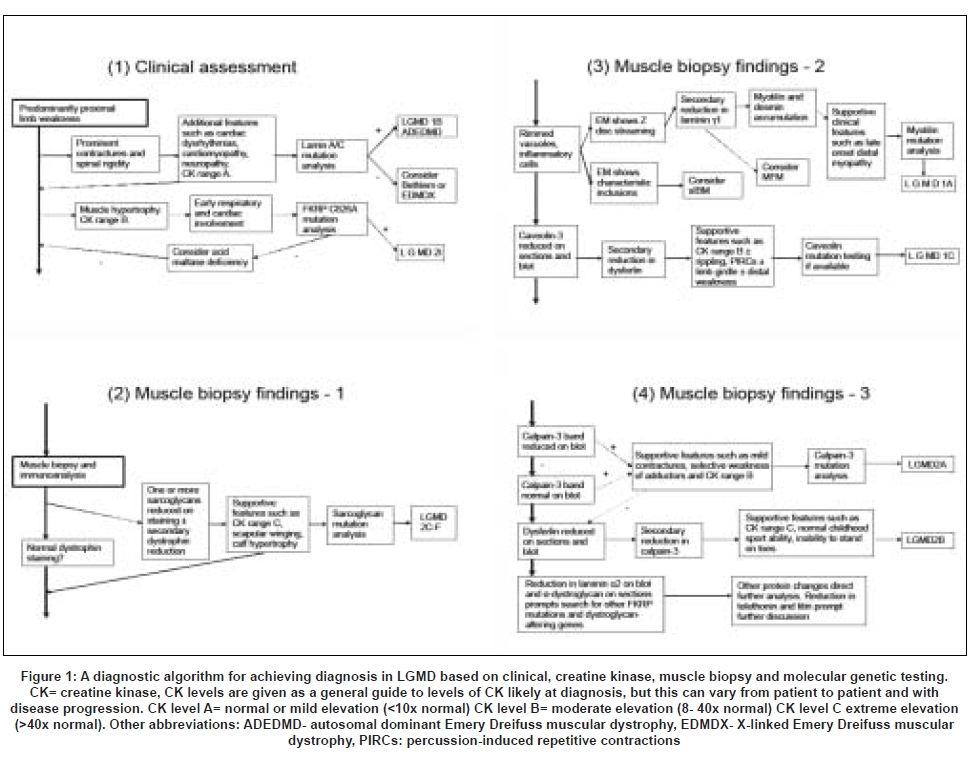
Combining information from the clinical and biopsy details therefore allows a diagnostic algorithm to be suggested and this is presented in [Figure 1]. In Part 1 of the algorithm the features which may suggest diagnosis of either LGMD2I or LGMD1B (neither of which can be definitively diagnosed by muscle biopsy) are described. Parts 2-4 describe the interpretation of the muscle biopsy findings together with clinical pointers which may lead to a suggestion of a genetic diagnosis which needs to be confirmed by testing. This diagnostic algorithm was previously published (EFNS guidelines).[10] DNA analysis is still the gold standard of diagnosis in LGMDs.
Autosomal dominant limb girdle muscular dystrophy
In most of the autosomal dominant AD-LGMD, the muscle biopsy is uninformative and shows an unspecific pattern that can either resemble a myopathy or a muscular dystrophy. No specific antibodies for immunohistochemical and Western Blot analysis are so far available for the AD-LGMD, with the exception of caveolin3.
LGMD2A
Immunoblotting has been an accepted test required for the diagnosis of LGMD2A.
However, there is variability in the quantity and function of calpain3 protein detected on immunoblots even for those patients with genetically confirmed LGMD2A. Secondary deficiencies of calpain3 have been described in several muscular dystrophies, including LGMD2B and LGMD2J. Moreover, a normal calpain3 expression on immunoblot has been observed in some patients in whom calpain3 mutations were proven [50],[51],[52],[53] and seems to be related to abnormal protein enzymatic activity.
LGMD2B
In LGMD2B, qualitative and quantitative protein analysis on muscle sections by immunostaining and Western Blot typically shows complete or partial absence of dysferlin , which seems to be predictive of mutations in the DYSF gene. [43] There may also be a secondary reduction in the expression of calpain3 and caveolin3. [52],[53],[54] The presence of inflammatory infiltrates is encountered at times and patients with dysferlinopathy may be misdiagnosed with polymyositis.
Sarcoglycanopathies
In sarcoglycanopathies the immunofluorescence analysis of muscle biopsy shows combined or more rarely isolated reduction of alpha, beta, gamma and delta sarcoglycans. A concomitant reduction of dystrophin and in same cases of dystroglycan expression can also be found, complicating the differential diagnosis with dystrophinopathies. In most cases immunofluorescence analysis or immunoblotting do not allow a reliable specific diagnosis because primary defects in a single sarcoglycan affect the expression of other sarcoglycans. A possible association between γ-sarcoglycan deficiency at the muscle biopsy and gene mutations has been reported [43] but so far, correct diagnosis requires genetic confirmation and analysis of several sarcoglycan genes may be necessary.
Alpha-dystroglycanopathies
The muscle biopsy immunostaining in patients with any alpha dystroglycanopathies usually shows a reduction in the dystroglycan expression if the antibody is directed against the carbohydrate moieties of alpha dystroglycan. The abnormal glycosylation pattern can also be detected on Western Blot. However, the commonly available antibodies can give unspecific results and the interpretation of the protein analysis may be difficult. Secondary reduction of laminin-alpha-2 on immunolabeling has been reported in most of the patients with alpha dystroglycanopathies and can be helpful to recognize these LGMDs. [55],[56]
Conclusion
Achieving a precise diagnosis in all patients is important to apply genetic counseling properly and to plan appropriate medical management. It will become increasingly important as new specific therapies are developed, and as trials of therapies in different types of LGMDs, including limited local gene therapy trials get under way. As diagnostic testing begins to be systematically applied in different countries, the epidemiology of these diseases can increasingly be understood, and a variable pattern of disease frequency will be elucidated. Therefore the future development of diagnostic testing and indeed therapeutic studies will need to take these regional differences into account. Particularly striking is the predominance of LGMD2I due to the common C826A mutation in Northern Europe, [18] compared to a relatively higher proportion of LGMD2A and LGMD2B cases in Southern Europe. A highly focused approach to LGMD diagnostics is already improving management and is going to become increasingly important in the future.[57]
References
| 1. | Hauser MA, Horrigan SK, Salmikangas P, Torian UM, Viles KD, Dancel R, et al. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum Mol Genet 2000;9:2141-7. Back to cited text no. 1 [PUBMED] [FULLTEXT] |
| 2. | Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology 2004;62:1363-71. Back to cited text no. 2 [PUBMED] [FULLTEXT] |
| 3. | Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, et al. Identification of mutations in the gene encoding lamin A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum Mol Genet 2000;9:1453-9. Back to cited text no. 3 [PUBMED] [FULLTEXT] |
| 4. | Benedetti S, Merlini L. Laminopathies: From the heart of the cell to the clinics. Curr Opin Neurol 2004;17:553-60. Back to cited text no. 4 [PUBMED] [FULLTEXT] |
| 5. | Hayashi T, Arimura T, Ueda K, Shibata H, Hohda S, Takahashi M, et al. Identification and functional analysis of a caveolin-3 mutation associated with familial hypertrophic cardiomyopathy. Biochem Biophys Res Commun 2004;313:178-84. Back to cited text no. 5 [PUBMED] [FULLTEXT] |
| 6. | Minetti C, Sotgia F, Bruno C, Scartezzini P, Broda P, Bado M, et al. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet 1998;18:365-8. Back to cited text no. 6 [PUBMED] [FULLTEXT] |
| 7. | Starling A, Kok F, Passos-Bueno MR, Vainzof M, Zatz M. A new form of autosomal dominant limb-girdle muscular dystrophy (LGMD1G) with progressive fingers and toes flexion limitation maps to chromosome 4p21. Eur J Hum Genet 2004;12:1033-40. Back to cited text no. 7 [PUBMED] [FULLTEXT] |
| 8. | Balci B, Uyanik G, Dincer P, Gross C, Willer T, Talim B, et al. An autosomal recessive limb-girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromusc Disord 2005;15:271-5. Back to cited text no. 8 [PUBMED] [FULLTEXT] |
| 9. | Nigro V. Molecular bases of autosomal recessive limb-girdle muscular dystrophies. Acta Myol 2003;22:35-42. Back to cited text no. 9 [PUBMED] |
| 10. | Norwood F, de Visser M, Eymard B, Lochmüller H, Bushby K. EFNS Guideline Task Force. EFNS guideline on diagnosis and management of limb girdle muscular dystrophies. Eur J Neurol 2007;14:1305-12. Back to cited text no. 10 |
| 11. | Straub V, Bushby K. The childhood limb-girdle muscular dystrophies. Semin Pediatr Neurol 2006;13:104-14. Back to cited text no. 11 [PUBMED] [FULLTEXT] |
| 12. | Olive M, Goldfarb LG, Shatunov A, Fischer D, Ferrer I. Myotilinopathy: Refining the clinical and myopathological phenotype. Brain 2005;128:2315-26. Back to cited text no. 12 |
| 13. | Decostre V, Ben Yaou R, Bonne G. Laminopathies affecting skeletal and cardiac muscles: Clinical and pathophysiological aspects. Acta Myol 2005;24:104-9. Back to cited text no. 13 [PUBMED] |
| 14. | Woodman SE, Sotgia F, Galbiati F, Minetti C, Lisanti MP. Caveolinopathies: Mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology 2004;62:538-43. Back to cited text no. 14 [PUBMED] [FULLTEXT] |
| 15. | Mercuri E, Bushby K, Ricci E, Birchall D, Pane M, Kinali M, et al. Muscle MRI findings in patients with limb girdle muscular dystrophy with calpain 3 deficiency (LGMD2A) and early contractures. Neuromuscul Disord 2005;15:164-71. Back to cited text no. 15 [PUBMED] [FULLTEXT] |
| 16. | Groen EJ, Charlton R, Barresi R, Anderson LV, Eagle M, Hudson J, et al. Analysis of the UK diagnostic strategy for limb girdle muscular dystrophy 2A. Brain 2007;130:3237-49. Back to cited text no. 16 [PUBMED] [FULLTEXT] |
| 17. | Saenz A, Leturcq F, Cobo AM, Poza JJ, Ferrer X, Otaegui D, et al. LGMD2A: Genotype-phenotype correlations based on a large mutational survey on the calpain 3 gene. Brain 2005;128:732-42. Back to cited text no. 17 |
| 18. | Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 1998;20:31-6. Back to cited text no. 18 [PUBMED] [FULLTEXT] |
| 19. | Nguyen K, Bassez G, Krahn M, Bernard R, Laforκt P, Labelle V, et al. Phenotypic study in 40 patients with dysferlin gene mutations: High frequency of atypical phenotypes. Arch Neurol 2007;64:1176-82. Back to cited text no. 19 |
| 20. | Klinge L, Dean AF, Kress W, Dixon P, Charlton R, Müller JS, et al. Late onset in dysferlinopathy widens the clinical spectrum. Neuromuscul Disord 2008;18:288-90. Back to cited text no. 20 |
| 21. | Okahashi S, Ogawa G, Suzuki M, Ogata K, Nishino I, Kawai M. Asymptomatic sporadic dysferlinopathy presenting with elevation of serum creatine kinase: Typical distribution of muscle involvement shown by MRI but not by CT. Intern Med 2008;47:305-7. Back to cited text no. 21 [PUBMED] [FULLTEXT] |
| 22. | Nguyen K, Bassez G, Bernard R, Krahn M, Labelle V, Figarella-Branger D, et al. Dysferlin mutations in LGMD2B, Miyoshi myopathy, and atypical dysferlinopathies. Hum Mutat 2005;26:165. Back to cited text no. 22 [PUBMED] [FULLTEXT] |
| 23. | Lim LE, Campbell KP. The sarcoglycan complex in limb-girdle muscular dystrophy. Curr Opin Neurol 1998;11:443-52. Back to cited text no. 23 [PUBMED] [FULLTEXT] |
| 24. | Eymard B, Romero NB, Leturcq F, Piccolo F, Carriι A, Jeanpierre M, et al. Primary adhalinopathy (alpha-sarcoglycanopathy): Clinical, pathologic, and genetic correlation in 20 patients with autosomal recessive muscular dystrophy. Neurology 1997;48:1227-34. Back to cited text no. 24 |
| 25. | Tsubata S, Bowles KR, Vatta M, Zintz C, Titus J, Muhonen L, et al. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest 2000;106:655-62. Back to cited text no. 25 [PUBMED] [FULLTEXT] |
| 26. | Moreira ES, Wiltshire TJ, Faulkner G, Nilforoushan A, Vainzof M, Suzuki OT, et al. Limb-girdle muscular dystrophy type 2G is caused by mutations in the gene encoding the sarcomeric protein telethonin. Nat Genet 2000;24:163-6. Back to cited text no. 26 [PUBMED] [FULLTEXT] |
| 27. | Frosk P, Weiler T, Nylen E, Sudha T, Greenberg CR, Morgan K, et al. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am J Hum Genet 2002;70:663-72. Back to cited text no. 27 [PUBMED] [FULLTEXT] |
| 28. | Saccone V, Palmieri M, Passamano L, Piluso G, Meroni G, Politano L, et al. Mutations that impair interaction properties of TRIM32 associated with limb-girdle muscular dystrophy 2H. Hum Mutat 2008;29:240-7. Back to cited text no. 28 |
| 29. | Schoser BG, Frosk P, Engel AG, Klutzny U, Lochmuller H, Wrogemann K. Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2H. Ann Neurol 2005;57:591-5. Back to cited text no. 29 |
| 30. | Brockington M, Yuva Y, Prandini P, Brown SC, Torelli S, Benson MA, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 2001;10:2851-9. Back to cited text no. 30 [PUBMED] [FULLTEXT] |
| 31. | Walter MC, Petersen JA, Stucka R, Fischer D, Schrφder R, Vorgerd M, et al. FKRP (826C>A) frequently causes limb-girdle muscular dystrophy in German patients. J Med Genet 2004;41:e50. Back to cited text no. 31 |
| 32. | Sveen ML, Schwartz M, Vissing J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann Neurol 2006;59:808-15. Back to cited text no. 32 [PUBMED] [FULLTEXT] |
| 33. | Darin N, Kroksmark AK, Ahlander AC, Moslemi AR, Oldfors A, Tulinius M. Inflammation and response to steroid treatment in limb-girdle muscular dystrophy 2I. Eur J Paediatr Neurol 2007;11:353-7. Back to cited text no. 33 [PUBMED] [FULLTEXT] |
| 34. | Balci B, Uyanik G, Dincer P, Gross C, Willer T, Talim B, et al. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromusc. Disord. 2005; 15: 271-275. Back to cited text no. 34 |
| 35. | Biancheri R, Falace A, Tessa A, Pedemonte M, Scapolan S, Cassandrini D, et al. POMT2 gene mutation in limb-girdle muscular dystrophy with inflammatory changes. Biochem Biophys Res Commun 2007;363:1033-7. Back to cited text no. 35 [PUBMED] [FULLTEXT] |
| 36. | Godfrey C, Escolar D, Brockington M, Clement EM, Mein R, Jimenez-Mallebrera C, et al. Fukutin gene mutations in steroid-responsive limb girdle muscular dystrophy. Ann Neurol 2006;60:603-10. Back to cited text no. 36 [PUBMED] [FULLTEXT] |
| 37. | Clement EM, Godfrey C, Tan J, Brockington M, Torelli S, Feng L, et al. Mild POMGnT1 mutations underlie a novel limb-girdle muscular dystrophy variant. Arch Neurol 2008;65:137-41. Back to cited text no. 37 [PUBMED] [FULLTEXT] |
| 38. | Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007;130:2725-35. Back to cited text no. 38 [PUBMED] [FULLTEXT] |
| 39. | Udd B, Vihola A, Sarparanta J, Richard I, Hackman P. Titinopathies and extension of the M-line mutation phenotype beyond distal myopathy and LGMD2J. Neurology 2005;64:636-42. Back to cited text no. 39 [PUBMED] [FULLTEXT] |
| 40. | Bushby K, Muntoni F, Bourke JP. 107th ENMC international workshop: The management of cardiac involvement in muscular dystrophy and myotonic dystrophy. 7th-9th June 2002, Naarden, The Netherlands. Neuromuscul Disord 2003;13:166-72. Back to cited text no. 40 |
| 41. | Wallgren-Pettersson C, Bushby K, Mellies U, Simonds A. 117th ENMC workshop: Ventilatory support in congenital neuromuscular disorders-congenital myopathies, congenital muscular dystrophies, congenital myotonic dystrophy and SMA (II) 4-6 April 2003, Naarden, The Netherlands. Neuromuscul Disord 2004;14:56-69. Back to cited text no. 41 |
| 42. | Zatz M, de Paula F, Starling A, Vainzof M. The 10 autosomal recessive limb-girdle muscular dystrophies. Neuromuscul Disord 2003;13:532-44. Back to cited text no. 42 [PUBMED] [FULLTEXT] |
| 43. | Guglieri M, Magri F, D'Angelo MG, Prelle A, Morandi L, Rodolico C, et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum Mutat 2008;29:258-66. Back to cited text no. 43 [PUBMED] [FULLTEXT] |
| 44. | Lo HP, Cooper ST, Evesson FJ, Seto JT, Chiotis M, Tay V, et al. Limb-girdle muscular dystrophy: Diagnostic evaluation, frequency and clues to pathogenesis. Neuromuscul Disord 2008;18:34-44. Back to cited text no. 44 [PUBMED] [FULLTEXT] |
| 45. | Sveen ML, Schwartz M, Vissing J. High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann Neurol 2006;59:808-15. Back to cited text no. 45 [PUBMED] [FULLTEXT] |
| 46. | Moore SA, Shilling CJ, Westra S, Wall C, Wicklund MP, Stolle C, et al. Limb-girdle muscular dystrophy in the United States. J Neuropathol Exp Neurol 2006;65:995-1003. Back to cited text no. 46 [PUBMED] [FULLTEXT] |
| 47. | van der Kooi AJ, Frankhuizen WS, Barth PG, Howeler CJ, Padberg GW, Spaans F, et al. Limb-girdle muscular dystrophy in the Netherlands: Gene defect identified in half the families. Neurology 2007;68:2125-8. Back to cited text no. 47 [PUBMED] [FULLTEXT] |
| 48. | Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited neuromuscular disorders: Past, present, and future. J Magn Reson Imaging 2007;25:433-40. Back to cited text no. 48 [PUBMED] [FULLTEXT] |
| 49. | Fischer D, Walter MC, Kesper K, Petersen JA, Aurino S, Nigro V, et al. Diagnostic value of muscle MRI in differentiating LGMD2I from other LGMDs. J Neurol 2005;252:538-47. Back to cited text no. 49 [PUBMED] [FULLTEXT] |
| 50. | De Paula F, Vainzof M, Passos-Bueno MR, de Cαssia M, Pavanello R, Matioli SR, et al. Clinical variability in calpainopathy: What makes the difference? Eur J Hum Genet 2002;10:825-32. Back to cited text no. 50 |
| 51. | Fulizio L, Nascimbeni AC, Spinazzi M, Piluso G, Ventriglia VM. Molecular diagnosis in LGMD2A: Mutation analysis or protein testing? Hum Mutat 2004;24:52-62. Back to cited text no. 51 |
| 52. | Anderson LV, Davison K, Moss JA, Richard I, Fardeau M, Tomι FM, et al. Characterization of monoclonal antibodies to calpain 3 and protein expression in muscle from patients with limb-girdle muscular dystrophy type 2A. Am J Pathol 1998;153:1169-79. Back to cited text no. 52 |
| 53. | Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995;81:27-40. Back to cited text no. 53 [PUBMED] [FULLTEXT] |
| 54. | Walter MC, Braun C, Vorgerd M, Poppe M, Thirion C, Schmidt C, et al. Variable reduction of caveolin-3 in patients with LGMD2B/MM. J Neurol 2003;250:1431-8. Back to cited text no. 54 [PUBMED] [FULLTEXT] |
| 55. | Poppe M, Cree L, Bourke J, Eagle M, Anderson LV, Birchall D, et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology 2003;60:1246-51. Back to cited text no. 55 [PUBMED] [FULLTEXT] |
| 56. | Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet 2001;69:1198-209. Back to cited text no. 56 [PUBMED] [FULLTEXT] |
| 57. | Jarry J, Rioux MF, Bolduc V, Robitaille Y, Khoury V, Thiffault I, et al . A novel autosomal recessive limb-girdle muscular dystrophy with quadriceps atrophy maps to 11p13-p12. Brain. 2007 Feb;130(Pt 2):368-80. Back to cited text no. 57 |
Copyright 2008 - Neurology India
The following images related to this document are available:
Photo images
[ni08077f1.jpg]
[ni08077t2a.jpg]
[ni08077t2b.jpg]
[ni08077t1.jpg]
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}