 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
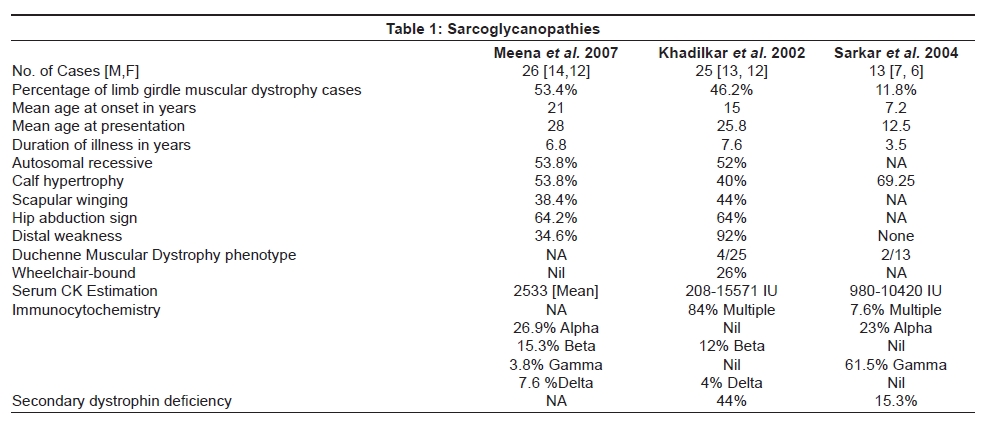
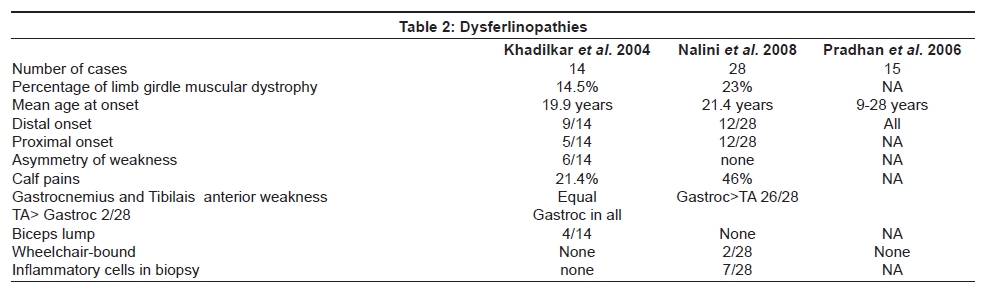
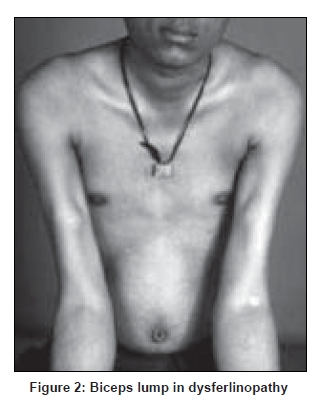
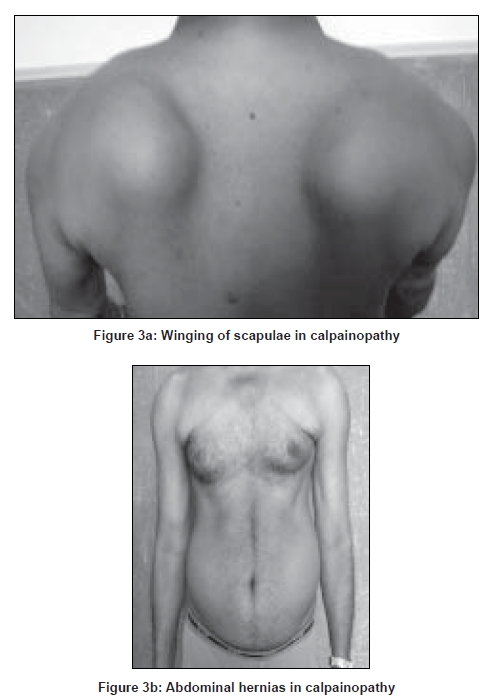
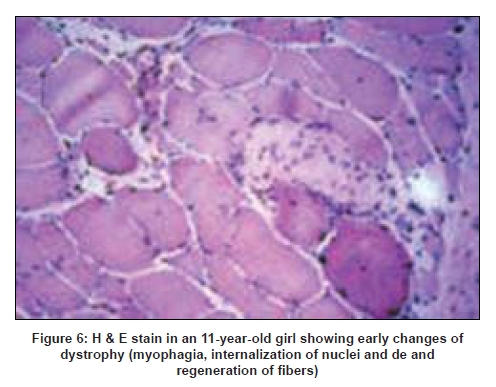
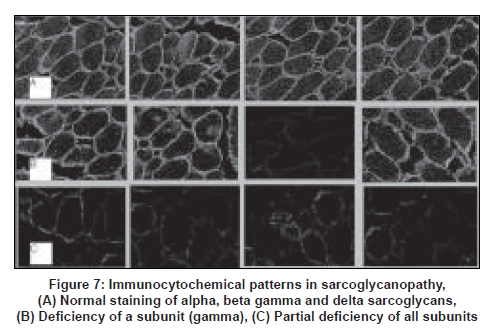
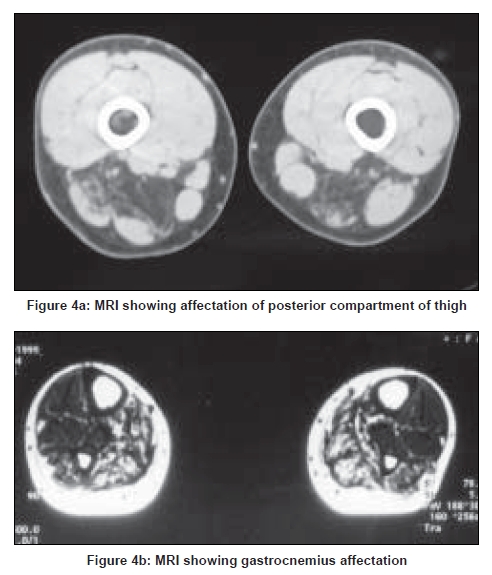
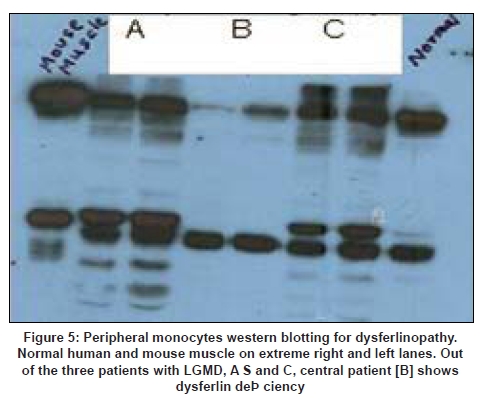
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 281-288 Review Article Limb girdle muscular dystrophies in India Khadilkar Satish V, Singh Rakesh K Department of Neurology, Grant Medical College and Sir JJ Group of Hospitals, Mumbai Date of Acceptance: 05-Aug-2008 Code Number: ni08078 Abstract The recent years have seen remarkable progress in the field of limb girdle muscular dystrophies (LGMDs) with the advances in immunocytochemistry and genetics. Based on this, many subgroups have emerged. Protein products and genes are getting defined and newer mechanisms of disease are being recognized. Limb girdle muscular dystrophies are common in India. The clinical material is plentiful, and from various centers in the country, phenotypes have been studied. With the help of immunocytochemistry, sarcoglycanopathies and dysferlinopathies have been studied. Genetic information on these subgroups is now beginning to emerge. The laboratory facilities are limited and available in select centers in large institutes. Establishment of genetic laboratories and sophisticated muscle pathology techniques will further elucidate the LGMDs in India.Keywords: Muscular dystrophies, limb girdle OR sarcoglycanopathy OR dysferlinopathy, India Evolution of Limb Girdle Muscular Dystrophies in India The historical descriptions of limb girdle muscular dystrophy (LGMD) date back to the late eighteenth century when Erb and Leyden-Mobius described patients having weakness which primarily involved the shoulder and the hip girdles respectively, spared the facial muscles, and were more benign than psuedohypertrophic form described by Duchenne . In 1951, Levison, [1] in his classification, documented for the first time, the autosomal recessive nature of LGMD. In 1953, Stevenson [2] introduced the term ′autosomal limb girdle muscular dystrophy′, for his non-Duchenne types with autosomal recessive inheritance. In 1954, Walton and Nattrass [3] defined LGMD as a distinct nosological entity. In the ensuing years, development of diagnostic methods made it evident that LGMD, as originally defined by Walton and Nattrass, was composed of a wide variety of neuromuscular disorders, for example, Kugelberg Wealander disease, polymyositis, endocrine myopathies, and some congenital and metabolic myopathies. This was when the diagnosis went in disrepute. Gardner Medwin omitted the term from the classification. Walton, in 1981 [4] altered his approach to this nosological problem. He retained the term limb girdle muscular dystrophy to designate a major category of dystrophy comprising four subtypes : 1) Autosomal recessive or sporadic (Erb, Leyden-Mobius) 2) Myopathy limited to quadriceps 3) Autosomal recessive muscular dystrophy in childhood and 4) Late onset Autosomal dominant type. The lack of uniformity of available classifications raised the issue as to what constituted the specific entity of LGMD. In the recent times, the advancement of molecular biology and immunological methods has seen the formation of subgroups based on molecular genetics and deficient components. Many types of autosomal recessive and dominant LGMDs based on genetic information or on protein deficiencies have been now described and newer types are being identified at a rapid pace. Since decades, across neuromuscular centers in India, dystrophies have been documented which were clearly distinct from the then well-described dystrophinopathies, myotonic disorders, fascio-scapulo humeral dystrophies and the likes. Initial observations on LGMD in India were made in 1975, when Srinivas, in a survey of 211 patients of myopathies seen over 25 years, diagnosed almost half the patients to have muscular dystrophy. Amongst them, 82 were Duchenne Muscular Dystrophy (DMD), 35 LGMD, five fascio-scapulo humeral dystrophies and 14 ocular-oculopharyngeal myopathies. [5] In a hospital-based study of 126 cases of muscular dystrophy by Mondkar and Bhabha, [6] 12 were designated as "girdle dystrophy" (three females) as they did not fit in Duchene or Becker muscular dystrophies. Four cases (three males) with affected sisters were labeled "Autosomal recessive dystrophy of childhood". In1998, Das published a large series of 1950 biopsy-proven myopathies, of which 535 were dystrophies. Among them, 29.2% were labeled as LGMDs, 5.6% severe dystrophies were seen in young girls, resembling DMD and 2.2% had autosomal recessive dystrophies in young boys. [7] In the more recent times, immunocytochemical stains have been used to study Indian patients with LGMDs and information based on immunostaining has emerged. Initial case reports of "Adhalinopathy" (alpha-sarcoglycanopathy) appeared in 2001. [8],[9],[10] This was followed up by case reports of beta and gamma-sarcoglycanoathy from pediatric centers in New Delhi. [11],[12] In 2002, the first series analyzing 25 cases of sarcoglycanopathy came from a neuromuscular center in Mumbai, where phenotypic features were studied in detail and some unique features were pointed out. [13],[14],[15] Soon, as more and more centers across the country got involved with immunostaining of myopathies, further reports, one from New Delhi in 2004, a study of 13 pediatric cases [16] and the other from Hyderabad in 2007, detailing 26 adult cases of sarcoglycanopathies became available. [17] Indeed, sarcoglycanopathy is the best characterized LGMD in India. The second type of LGMD to be immunocharacterized in India is dysferlinopathy. The initial series of 14 Indian patients with dysferlinopathy from Mumbai was published in 2004. [18] The second study comprising nine cases was recently reported from northern India by Pradhan in 2008. [19] A third series comprising 28 patients from south India has been made available in 2008. [20] Genetic evaluation of LGMDs is only recently being reported and is essentially related to small samples. Due to the complexity of LGMD genetics, it seems to have lagged behind other common neuromuscular disorders like spinomuscular atrophy [21] and dystrophinopathies. Prevalance Due to the heterogeneity of LGMD and the lack of diagnostic specificity, estimates of prevalence for all forms of LGMD have ranged from 1 per 14500 to 1 per 123000 populations. In the Indian setting, it is difficult to know the exact prevalence. In a hospital-based survey in a neuromuscular clinic in Mumbai, approximately one-fourth of all patients had LGMD. In adulthood, LGMD formed the most common diagnosis in the group of muscular dystrophies. The series by Das and Mondkar also quote similar figures. This is interesting as LGMDs were far commoner than myotonic disorders, which form the most common adulthood myopathies in some populations. However, given the diversity of ethnicity and genetic background of Indian populations, further systematic and multi-center information is needed to make a useful assessment of the prevalence of LGMD.Inheritance Patterns India is a vastly populous country with diverse cultural conglomerates. In general, autosomal recessive forms of LGMD outnumber the dominant forms. In some communities in India, consanguinity is customary and may be leading to increase in autosomal recessive transmission of LGMDs. Another noteworthy point is the apparent lack of family history in patients with LGMDs. In hospital-based data of 15 years from Mumbai, only 30% patients with LGMD had family history of the disease. This is perplexing and the reason why we do not actually pick up the autosomal recessive states in a proportion of our patients is possibly due to the inadequate ancestoral information given by them and lack of available family members for examination. In the neuromuscular center at Mumbai, social worker inputs in getting together the family members has increased the yield of the number of familial cases to some extent. Autosomal dominant LGMDs are seen less often. In one study at Mumbai, 37 recessive and five dominant cases were encountered. [14] At times, due to deeply inbred population, as seen in certain communities in our country, pseudo-dominance may be encountered.Phenotypic Features LGMD has been prospectively analyzed in many centers across the country. As mentioned earlier, it is the most common adult onset muscular dystrophy encountered in our country. Thanks to a large number of cases, robust clinical data has been generated. Particular attention has been devoted to the phenotype. As a result, interesting signs were described by authors. [15],[19] Among LGMDs in India, sarcoglycanopathy has been most extensively studied followed by dysferlinopathy. With time, as further facilities are obtained, information on other subtypes is expected to emerge. Sarcoglycanopathies The most commonly noted phenotype of sarcoglycanopathies has been of hip-girdle weakness predominantly involving thigh adductors and knee flexors. The abductors are not much affected till late in the illness. Hence patients do not waddle. In the thigh, hamstrings and quadriceps are both affected and the relative severity is variable. The upper limbs are less involved and the biceps tend to be weaker than the deltoid muscle. Thus in certain ways, the clinical involvement of sarcoglycanopathies is different from Becker muscular dystrophy wherein the hip abductors and quadriceps are affected more severely. "Hip-abduction sign" (splaying of thighs while getting up from squatting position) is the result of differential weakness between thigh adductors and abductors, the latter being strong. [15] The consistency with which this sign is observed is noteworthy. Some patients learn tricks to prevent splaying of thighs by bringing the knees together and supporting the outer aspects of the thighs with hands. The sign comes to the clinical notice early in the disease course due to the prevalent floor toilet system in India. Some patients report intriguing initial complaints like inability to move the leg from the accelerator to the brake, thighs splaying while performing sit-ups and also while getting up from the ground. Although the sign is a useful clue while evaluating a dystrophy case, it is not specific for a subtype of LGMDs and is seen beyond sarcoglycanopathies. It actually denotes stronger abductors and weaker adductors and has a differential diagnosis. Distal weakness was consistently noted in adult onset sarcoglycanopathies from Mumbai. It was moderate in severity. This has not been commonly mentioned in world literature. Sarkar et al. , [16] did not notice any distal weakness in their mostly pediatric case series. Phenotypic variability is quite common in LGMDs. Severe weakness in childhood, which is SCARMD, is usually the result of alpha and gamma-sarcoglycan [SG] mutations worldwide. [22],[23] In the Indian series it was secondary to almost all SGs, [15],[16],[21] based on immunodiagnosis. One of our families, a brother and sister sib ship with SCARMD phenotype, [24] did not show deletions in any of the known sarcoglycan genes. This is in accordance with the fact that SCARMD phenotype can result from other mutations. [25] When female members are not affected, the differentiation from DMD becomes difficult. Patients with DMD are more likely to have significant heart involvement and mental sub-normality than LGMD. The severity of clinical presentation varies greatly in the group of sarcoglycanopathies. At one end there is the severe DMD phenotype with early disability and reduced life expectancy. On the other hand, milder forms allowing ambulation till the third and fourth decades are seen. The genotype-phenotype correlation is complex and all the genotypes can result in variable phenotypes. However, alpha and gamma sarcoglycanopathies tend to have severe presentation. As seen from , milder presentations of sarcoglycanopathies are not uncommon in India. Dysferlinopathies The series by Pradhan et al. , [24] focuses on the clinical features of patients showing dysferlin deficiency on immunostaining. A specific appearance of the shoulder girdle, simulating calf head on trophy, has been noted in patients with Miyoshi myopathy. Table 2 compares the three available series of dysferlinopathies from India. As can be seen from these three papers, disability tends to be mild in dysferlinopathy patients and ambulation is maintained for a long time in the illness. Selectivity of involvement muscle groups is a hallmark of dystrophy. Such selective wasting within the same muscle results in a unique appearances of muscle mass. ′Lumps and bumps′ of quadriceps in sarcoglycanopathy [13] Figure 1, ′Biceps lump [18] Figure 2, ′diamond sign′ (prominence of anterolateral aspect of thigh on partial contraction of quadriceps) in dysferlinopathy, [19] ′calf-head sign′ in Miyoshi myopathy [26] and valley sign [27] are some examples. Biceps lump is believed to be the result of partial involvement of biceps muscle. It was initially described by Fardeu and colleagues from Paris and is believed to be seen most often in dysferlinopathy. Valley sign, a depression created by selective wasting of posterior axillary fold muscles bordered by relatively preserved deltoid superiorly and infraspinatus inferiorly, described by Pradhan mainly in DMD, [27] has been seen in some patients with LGMDs. [17] Symptomatic cardiac involvement like cardiomyopathy is relatively uncommon in autosomal recessive LGMDs but patients tend to have subclinical abnormality of ECG and echocardiograms, especially T-wave inversions, left ventricular hypertrophy and right bundle branch block. [17],[28] In contrast, a larger proportion of autosomal dominant LGMD patients have significant cardiac disease, like cardiomyopathy or conduction blocks. [18] Central nervous system abnormalities, especially mental retardation have been recently described in LGMD2K and 2I, which occur due to mutations in the POMT1 and fukitin proteins respectively. [29],[30],[31] This protein is also expressed in the brain and hence mental sub-normality results. In our series, we encountered an AR-SCARMD phenotype in a brother and sister with mental retardation and generalized chorea. Proband had normal dystrophin and sarcoglycan staining. [24] All known sarcoglycan genes were normal. Immunoblot for alpha-dystroglycan is not yet available with us. One unpublished family of two siblings with mental sub-normality and proximal and distal weakness having POMT1 gene abnormality is being followed up at Mumbai. Description of calpainopathy is currently lacking in the Indian literature, mainly due to the unavailability of immunoblotting techniques. Phenotype suggestive of calpainopathy has been seen. In our series of LGMD at Mumbai, we have come across five sporadic patients with the typical phenotype of severe scapular winging without facial weakness [Figure 3a, b]. They had normal sarcoglycan and dysferlin staining, but had not as yet undergone calpain immunoblotting. Autosomal dominant LGMDs are less common. We could identify five cases. The most distinct feature was the severe cardiac ailment noted in four of them. In fact one of our patients was referred from the cardiology department where he was admitted with cardiomyopathy. In a family of three brothers and three affected generations, the index case had cardiac pacemaker implanted for heart block, the other two brothers had muscular dystrophy and cardiac abnormalities. This family very likely represents autosomal dominant LGMD of the laminopathy type. Diagnostic Aspects of LGMD in India In the diagnostic workup of suspected LGMD in India, after biochemistry and electrophysiology tests, muscle biopsy is often performed. It is done to establish the dystrophic nature of the process and to rule out alternative causes of muscle weakness. The chosen muscle is the one with moderate weakness. Histology with the conventional H&E staining helps to the establish the nature of the dystrophic process by demonstrating the variation of fiber size, internalization of nuclei, splitting of fibers, myonecrosis, myophagia and changes in connective tissue. Figure 6 shows early dystrophic changes in an 11-year-old girl with sarcoglycanopathy. Presence of inflammatory cells has been noted in various types of LGMDs, especially in dysferlinopathy. The inflammatory cells often lead to an erroneous diagnosis of inflammatory myopathy and hence evaluation of all changes in the biopsy is important. Alone, histology has a limited role and needs be followed up with immunocytochemistry. Immunocytochemical analysis of the frozen muscle tissue using monoclonal antibodies for dystrophin, sarcoglycans, dysferlin and merosin is now regularly performed in dedicated neuromuscular centers across India. The results of sarcoglycan staining have been variable among different centers in India. While the study from Mumbai found multiple SG deficiency as the most common pattern, others had either gamma [16] or alpha SG deficiency. [17] Reasons for this discrepancy could be different age groups of patients, early or late stages of the disease or speculatively, even ethnicity of the studied population. As yet, it is not possible to know which type of sarcoglycan deficiency is the most prevalent in India. Figure 7 shows various patterns of abnormalities of sarcoglycan immunostaining. It needs to be pointed out here that immunocytochemistry has its own inherent limitations. The sarcoglycan hypothesis forwarded by Mizuno [32] states that if one of the SG subunits is absent, the whole complex gets deficient on the muscle membrane. The implication is that on staining patterns alone without genetic studies, it is difficult to know the primary abnormality. Also, no immunostaining pattern is considered specific for a particular sarcoglycan deficiency. To make matters worse, there is complex interaction of sarcoglycans with dystrophin and dysferlin and other stains, which can be secondarily deficient. Immunoblotting techniques can further improve the diagnostic accuracy by quantitating the protein. The differences in the accuracy of quantitative and qualitative assessment are well known. Moreover some subgroups like calpainopathy cannot be confirmed without western blotting. Hence for a confident diagnosis of an LGMD subtype it is preferable to study all available immunostains, quantitate them by western blotting and also study the genes. Magnetic resonance imaging (MRI) is noninvasive and easily available in our country. It clearly demonstrates the selectivity of muscle weakness [Figure 4 a, b]. At times, in a difficult case, choice of muscle for biopsy can be guided by the MRI findings. Biochemical characterization of LGMD muscle tissue was done by Sharma and Sarkar [33] by MRI-spectroscopy. They found reduced choline, glucose and glutamine in the affected muscles. In dysferlinopathy, peripheral blood can be studied for the dysferlin western blotting to help the diagnosis Figure 5. However, this does not completely obviate the need for muscle biopsy and genetic tests. Genetics of Sarcoglycanopathies in India Evaluation of a Patient with LGMD Approach to a patient with limb girdle weakness entails detailed history and clinical examination. It pays to take in-depth family history and examine the family members, to narrow down the differential diagnosis. Particular attention is paid to the phenotype, which gives some clue to the type of LGMD. Electrophysiology and laboratory tests including CPK, toxic and metabolic screen are done as part of diagnostic workup. To distinguish LGMD from dystrophinopathy can be a problem, especially in a young boy with severe weakness (SCARMD phenotype). Dystrophin immunostaining and Polymerase chain reaction (PCR)-based dystrophin gene analysis are available in many centers across India, to help this differential diagnosis. The broad classification of a patient in the LGMD group is essentially clinical and is based on the common features of transmission and phenotypic expression. However, focusing on a subtype of LGMD is not clinically feasible given the phenotype-genotype heterogeneity. The subgrouping is relevant to the genetic counseling issues and to an extent, may be a predictor of the course of illness and the cardiac involvement as well. Establishing the specific subtype of LGMD in a given patient needs detailed immunocytochemical analysis for various muscle proteins. Demonstration of partial or complete deficiencies of a particular protein in immunostaining should ideally be followed up with mutation studies of the corresponding gene. It is important to realize that immunocytochemistry has limitations and it is difficult to base the gene search based only on immunocytochemical findings. Conclusions Limb girdle muscular dystrophies form an important and common group of myopathies in India. Only two subgroups, sarcoglycanopathies and dysferlinopathies have been characterized up to a point. The diagnostic aspects are still evolving and genetic information is lacking. With wider availability of tests, more information about this group of myopathies will come to the fore.References
Copyright 2008 - Neurology India The following images related to this document are available:Photo images[ni08078f4.jpg] [ni08078f2.jpg] [ni08078f5.jpg] [ni08078t1.jpg] [ni08078f6.jpg] [ni08078f3.jpg] [ni08078t2.jpg] [ni08078f1.jpg] [ni08078f7.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}