 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
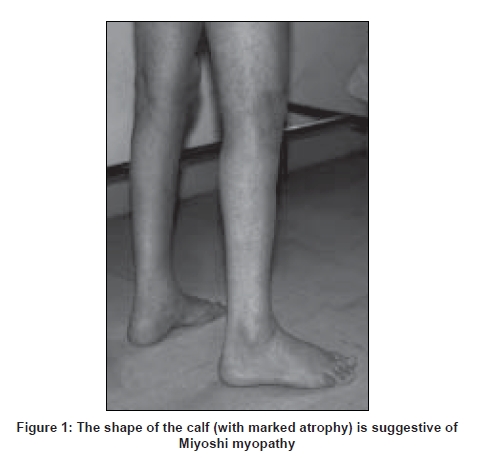
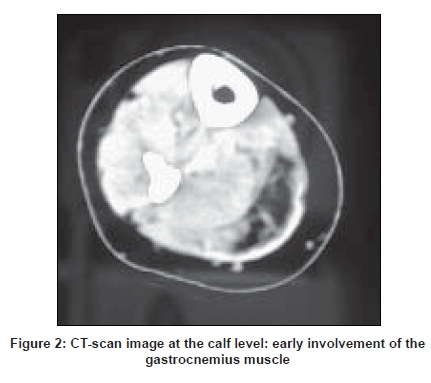
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 289-297 Review Article Dysferlinopathies Urtizberea J Andoni, Bassez Guillaume, Leturcq France, Nguyen Karine, Krahn Martin, Levy Nicolas Assistance Publique Hopitaux de Paris, Hopital Marin, BP40139, 64700 Hendaye Date of Acceptance: 08-Aug-2008 Code Number: ni08079 Abstract Dysferlinopathies encompass a large variety of neuromuscular diseases characterized by the absence of dysferlin in skeletal muscle and an autosomal recessive mode of inheritance. So far, three main phenotypes have been reported: Miyoshi myopathy (MM), limb girdle muscular dystrophy type 2B (LGMD 2B), and distal myopathy with anterior tibial onset (DMAT). A growing number of clinical variants have recently been described with a much wider range of symptoms and onset. Although rare, dysferlinopathies can account for up to 30% of progressive recessive muscular dystrophies in certain geographical areas, notably in the Middle East and the Indian subcontinent. Dysferlin is a large protein involved in membrane repair and vesicle trafficking and interacts probably with important immunological pathways. New insights in its pathophysiology may result in innovative therapies in the near future. Keywords: Distal myopathy with anterior tibial onset, dysferlinopathies, dysferlin, limb girdle muscular dystrophy, LGMD 2B, Miyoshi myopathy, muscular dystrophy, mini-dysferlin Historical Landmarks The term ′dysferlinopathy′ was coined in 1999 by Bushby after Miyoshi myopathy (MM) and limb girdle muscular dystrophy type 2B (LGMD 2B) were found to be allelic disorders. [1] Nowadays, it corresponds to the various clinical phenotypes related to a complete or partial absence of dysferlin. Historically, the first phenotype of dysferlinopathy to be described was the one reported by Miyoshi, a Japanese physician, in 1967 and subsequently in 1986. [2],[3] In Miyoshi′s original publication, four patients from two consanguineous families presented with recessively inherited late-onset distal myopathy associated with clear-cut muscular dystrophy and significantly elevated creatine Kinase (CK) levels. This phenotype was called Miyoshi myopathy (MM). Long thought to be confined to Japan, the disease was soon reported in Europe and elsewhere thereafter. In 1995, Bejaoui, from the Boston group, mapped the MM locus to chromosome 2p14-p12 by studying 12 informative families, five of which were consanguineous. [4] In 1996, Weiler et al., reported the coexistence, in a large Canadian aboriginal family, of MM on the one hand and of a condition mimicking LGMD on the other. [5] Similar findings had been reported earlier by Mahjneh in a large kindred from Palestine. [6] Limb girdle muscular dystrophy is the generic name for a heterogeneous group of neuromuscular disorders inherited either dominantly of recessively. They are characterized by muscle wasting and atrophy mostly in the shoulder and pelvic girdle but with a quite variable degree of severity and disease course. The vast majority of LMGDs are recessively inherited. In 1994, the first gene to be cloned in a recessive LGMD was unraveled by Richard. [7] It rapidly became obvious that many other recessive genes were involved in that group of patients. The same year, the Newcastle team showed that another group of LGMD families, one of which was located in Palestine, was particularly informative for linkage studies [6] and actually mapped to another locus, on chromosome 2p. [8] The LGMD international consortium agreed in Naarden in 1995 to name the different LGMD loci based on chronological order and mode of inheritance. As a result, calpain deficiency was termed LGMD 2A and the other locus became LGMD 2B (the suffix "2" is meant to point out the recessive forms). In 1998, after several successful attempts to refine the interval of the MM/LGMD2B locus to the 2p13 region, two independent laboratories (located in Newcastle and Boston respectively) cloned the dysferlin gene and clearly demonstrated that the two disorders (MM and LGMD 2B) were allelic and caused by the same gene defect (DYSF). [9],[10] Clinical Phenotypes Miyoshi myopathy (OMIM # 254310) Limb girdle muscular dystrophy type 2B (OMIM #253601) Distal myopathy with onset in tibialis anterior (OMIM # 606768) Other clinical variants In other instances, the disease manifests as a pseudometabolic disorder with exercise intolerance, myalgias and cramps, but without myoglobinuria. Isolated and long-lasting hyperCKemia has also been reported. Elevated CK levels and minimal clinical manifestations have also been anecdotally described in a few heterozygotes. [20] Interestingly, many authors reported that onset of disease can be quite asymmetrical, unilateral, with or without transient calf myalgia and swelling. In some unusual cases, it inappropriately led to the diagnosis of thrombophlebitis. A case of dysferlinopathy with choreic movements has been described only once and may just be coincidental. [21] In elderly people, spinal muscle degeneration, with or without camptocormia can also be a presenting symptom. [22] Several comprehensive surveys have stressed the importance of these clinical variants and advocate for a broader view as far as the spectrum of dysferlinopathies is concerned. [18],[23],[24] For instance, age at onset is more variable than originally thought, with some patients remaining asymptomatic till 60 or 70 years of age, and conversely, patients becoming symptomatic in their early teens. The association between dysferlinopathy and cardiomyopathy is still a debated issue. [25] Some authors argue for its anecdotal nature while others, putting forward the evidence accumulated from the murine animal model of dysferlinopathy (SJL mouse), think that this cardiac complication might be overlooked in patients. [26],[27] Recently, a group from Berlin, Germany, reported two out of six LGMD/MM unrelated patients, who developed overt cardiac disease without any correlation with the severity of their skeletal muscle phenotype. This combination may exist in other patients but this has not been fully investigated so far. Such a working hypothesis fully justifies prospective heart studies in patients with genotypically proven dysferlinopathy. Distribution of Phenotypes While DMAT remains rather uncommon and was reported mainly in Spain and Japan, [15],[28] MM and LGMD 2B seem to be equally frequent in the overall patient population with dysferlinopathy (personal data). Other clinical variants may account for 20-30% of the total, based on the different available worldwide studies. More importantly, at least two, more rarely three or more, phenotypes (MM, LGMD 2B, DMAT, others) can coexist within the same family or pedigree as reported on several occasions, first in Canada [5] and elsewhere. [18],[29],[30],[31],[32] So far, no satisfactory explanation has been given to account for this unusual occurrence. Modifier genes or environmental factors are likely to be involved. Differential Diagnosis Misdiagnosis is commonplace in patients with primary dysferlinopathy. Worse, it can lead to unnecessary and potentially hazardous therapeutic interventions such as long-term oral administration of corticosteroids or immunosuppressors. In a couple of retrospective studies, close to 25% of LGMD 2B/MM patients had initially been diagnosed with polymyositis, one of the most common dysimmune inflammatory myopathy in adults. In dysferlinopathy, inflammatory changes in muscle are sometimes so florid that they can mislead the pathologist as well as the clinician. Moreover, CK levels are equally elevated in both conditions, a finding which also adds to the confusion. When faced with similar challenges, it is best to systematically perform additional immunostains in order to differentiate the two conditions. In dysferlinopathies, the HLA-Class I complex is not over-expressed. [33] In any event, the most critical clue will be generated by dysferlin immunocytochemistry and immunoblotting as mentioned earlier. When CK levels are less informative (at end-stages of disease, for instance), alternative forms of distal myopathy may need to be differentiated. Dysferlinopathy remains by far the most frequent cause of autosomal recessive distal myopathy. [34],[35] Nonaka distal myopathy is sometimes an option, especially if onset is in the anterior compartment of the lower leg. Age of onset and disease progression can overlap with MM. However, the presence of numerous rimmed vacuoles in muscle is a distinctive feature; even though such vacuoles have occasionally been reported in primary dysferlinopathy. Except in large inbred kindred where pseudo-dominance can be observed and in sporadic cases of MM, the autosomal dominant forms of distal myopathy (Welander myopathy, Udd-Markesbery-Griggs myopathy, Laing myopathy) are much less relevant to the differential diagnosis. By taking into account the topography of the predominantly affected muscles (in the upper limbs in Welander disease) or the age at onset (in childhood in Laing myopathy, in rather late adulthood in Markesbery-Griggs myopathy), a definite diagnosis is often reached. But once again, immunostaining of dysferlin in muscle or monocytes is essential in that context and will be quite discriminating. With the exception of Welander myopathy, genetic markers are now available for most of these variants of distal myopathies (GNE mutations in Nonaka myopathy, titin mutations in Udd myopathy, ZASP mutations in Markesbery-Griggs myopathy and MYH7 mutations in Laing distal myopathy respectively). When dysferlinopathy presents as LGMD, the two alternate diagnoses are LGMD 2A and LGMD 1C, precisely because both entities can result in secondary dysferlinopathy with significant signal reduction either of calpain or of caveolin-3. Other forms (sarcoglycanopathies and FKRP-deficiencies) are easier to rule out. Charcot-Marie-Tooth disease and other neurogenic conditions also form a differential diagnosis. Sparing of the extensor digitorum brevis muscle in dysferlinopathy is usually a distinctive feature. The muscle tends to be affected in most neurogenic conditions. Epidemiology In the absence of a comprehensive international patients′ registry, the overall prevalence of dysferlinopathies is still hard to estimate. Additionally, figures vary from one place to another. As recessive disorders, MM and LGMD 2B are commonly reported in communities or geographical areas in which endogamy is high. It is true notably in Maghreb, Israel, Saudi Arabia, Iran and the Indian subcontinent where cases with dysferlinopathy are many. In these areas, dysferlinopathies are now emerging as the second most common cause of autosomal recessive LGMD in adults after calpain deficiency (LGMD 2A). In less consanguineous areas, such as the USA, they can still account for 15% of autosomal recessive LGMD. [36] Population-specific mutations and founder effects have been described in communities such as the Libyan Jews and more recently in the so-called Mountain Jews originating from the Caucasus region. [37],[38] Similarly, recurrent mutations in supposedly unrelated families have been picked in Japan, Italy and Spain. [32],[39] A founder effect is also likely in a cluster of Acadian/Cajun patients with proven dysferlinopathy settled both in Nova-Scotia and in Louisiana (personal unpublished data). It is somewhat difficult to distinguish a recurrent mutation in a large extended pedigree from a genuine founder effect within a strongly inbred community. Interestingly, no such mutational hotspots have been reported as yet in any of the three Maghrebian countries (Morocco, Algeria, and Tunisia) despite the equally high prevalence of primary dysferlinopathy in this area. The ongoing registration of all patients worldwide would certainly help clarify this point. Diagnosis With the advent of routine immunocytochemistry and targeted molecular genetics, diagnosing dysferlinopathy has become an easier task than before. However, a number of pitfalls exist for which several complementary approaches should be considered. Creatine Kinase (CK) levels Electromyographic (EMG) studies Muscle imaging Muscle imaging of the upper limbs may also be valuable but is technically more challenging to perform and interpret. In DMAT, muscle wasting is initially noted in the tibialis anterior muscle but extends to the gastrocnemius over time, generally in a couple of years. Muscle histology Dysferlin is recognized at the sarcolemma by at least two commercially available monoclonal antibodies (Hamlet-1 and Hamlet-2 antibodies are directed against two far-apart epitopes of dysferlin). In muscle, such antibodies detect the vast majority of dysferlin deficiencies. [18],[23],[41] It is noteworthy that they equally work nowadays with peroxydase and in immunofluorescence. There is a wide range of stain intensities between a complete absence of signal and a mere weak immunostain. The interpretation of the weak or slightly reduced immunostains remains challenging as secondary dysferlinopathies have been described in other muscular dystrophies. At the ultrastructural level, alterations of the membrane are present such as small tears and accumulation of subsarcolemmal vesicles and vacuoles, but such anomalies are not routinely investigated. [42] Western blotting A complementary assay consists of measuring the dysferlin content in an enriched pool of monocytes called CD-14+. The test is robust and simply requires blood sampling and shipping to an appropriate laboratory within 48 h. The sensitivity and sensibility of both tests are quite acceptable. The blood-based assay on monocytes is commercially available through Athena Diagnostics. Primary versus secondary dysferlinopathies DNA studies When faced with an MM phenotype without any pathogenic mutations in the DYSF gene, the hypothesis of a genetic heterogeneity in MM may arise. As a matter of fact, a few families with a true Miyoshi phenotype do not exhibit any dysferlin deficiency as reported earlier [46] and do not map to chromosome 2p. Some of them have even been mapped to another locus (Chromosome 10). [47] In a couple of these non-dysferlin patients, a comparable defect in membrane repair mechanisms has also been documented. [48] Genetic Counseling By definition, all cases of dysferlinopathies are transmitted following an autosomal recessive mode of inheritance. A pseudo-dominant transmission is occasionally observed in very endogamic communities with multiple consanguineous loops. The risk of recurrence among sibships is usually of 25%. Consanguinity does not raise the figure per se but increases the probability of two heterozygotes (healthy carriers) belonging to the same inbred community marrying and therefore giveing birth to an affected child. Given the relative mildness observed in the majority of dysferlinopathies, prenatal testing is seldom offered even if the gene defects are accurately known in a given patient. Carrier testing can be considered in inbred populations as long as a founder mutation has been found. Such preventive measures have been proposed already in Libyan and Caucasian Jews living in Israel. Genetic counseling sometimes turns out to be complex due to the fact that the different phenotypes, with variable disease progression, can coexist in the same sibship. At this point, it is impossible to predict whether the patient will develop as an MM or as an LGMD. Diagnostic Algorithm The preliminary clinical ascertainment is crucial and generally provides relevant clues to the diagnosis of dysferlinopathy. As mentioned above, a ′distal touch′ in the clinical picture is quite suggestive and is often confirmed by calf muscle imaging. A total absence of dysferlin in monocytes and/or in muscle is a key element for the positive diagnosis. Even better is to detect a homozygous mutation in the DYSF gene. This is seen in most consanguineous pedigrees. The fact of not finding out a second heterozygous mutation in the DYSF gene is not enough in itself to reject the diagnosis of dysferlinopathy. On the other hand, when the dysferlin immunostain shows some remnants of the protein, one has to be cautious, especially if the mutation screening turns out to be negative. A secondary dysferlinopathy is therefore a working hypothesis and this should prompt re-appraisal of the clinical and histological data. Pathophysiology Dysferlin is a large protein (230 kDa), composed of 2080 amino-acids with a C-terminal transmembrane domain and six calcium-binding C2 domains. Dysferlin is located mainly at the muscle membrane in adult muscle and at the T-tubule system in early development. [49],[50],[51] Dysferlin is widely expressed not only in skeletal muscle and cardiomyocytes, but also in non-myofibers such as monocytes (in CD14+ T cells, in particular). Dysferlin is a sarcolemmal protein sharing homology with the sperm vesicle fusion protein FER-1 that mediates fusion of intracellular vesicles with the spermatid plasma membrane. Dysferlin has been shown to be involved in sarcolemmal repair. [52],[53],[54],[55] Dysferlin-containing vesicles fuse to form a "membrane patch". The patch would then be added to the membrane disruption site for resealing. Dysferlin belongs to the larger family of ′ferlins′ that include myoferlin, otoferlin and others (FER1L4, FER1L5, and FER1L6). Dysferlin may also play a role in the central nervous system, notably at the level of the blood-brain barrier. [56] Two naturally-occurring mouse mutants are used to study the putative functions of dysferlin and the natural course of disease. The SJL/J mice carry a spontaneous splice-site mutation resulting in a 171-bp in-frame deletion removing 57 amino acids in the C2-E domain of the dysferlin protein. [57] Interestingly, SJL/J mice have been used extensively as a model for auto-immune disease. Another mouse, the A/J mouse, has a unique ETn retrotransposon insertion within intron 4 of the DYSF gene. Other transgenic mice are in preparation to further understand the role of the different parts of the protein in muscle physiology. Detailed interactions of dysferlin with other proteins are under investigation. While dysferlin co-localizes with annexin A1 and A2 at the sarcolemma, it also interacts with caveolin-3, affixin and AHNAK nucleoprotein. [58],[59],[60] In parallel, it has been shown that dysferlin also results in downregulation of the complement-protection molecule CD-55. These immunological findings coupled with the high degree of inflammation observed in early dysferlinopathy is an interesting avenue to explore. Indeed, macrophages (the circulating version of monocytes) exhibit an overaggressive activity in muscle fibers and may therefore contribute significantly to disease onset and progression. [61] Genotype-Phenotype Correlations So far, the relationship between phenotype and genotype in dysferlinopathy has remained extremely loose. Different mutations in DYSF have varying effects on protein expression. [18],[62],[63] As a result, the type of mutation does not correlate with phenotypic severity, and the same mutation has been found to be associated with a wide inter- and intra-familial variation in clinical phenotype. The best example is given by the concomitant observation of three or more clinical phenotypes (MM, LGMD, DMAT) within the same sibship or kindred. Even, the amount of residual dysferlin, if any, in muscle rarely correlates to the disease severity. It is obvious now that other modifying factors, or genetic and environmental nature, interfere.Dysferlinopathy in the Indian Context Dysferlinopathies are definitely ubiquitous conditions. [18],[23],[35],[64],[65] They have been observed in all ethnic groups, but have some propensity to occur in endogamic communities. Whether or not this group of disorders is more prevalent in India than in the Western world remains to be proved, but it seems highly plausible. A growing number of Indian teams have reported series of patients with dysferlinopathy. [12],[13],[66],[67] Immunocytochemistry for dysferlin has only been introduced in India recently, most exclusively in academic centers dedicated to myology (via Hamlet and Hamlet-2 antibodies marketed by Novacastra). Muscle Western blotting, though, is still at a preliminary stage and mastered only by a couple of laboratories across the country. The dysferlin assay on monocytes had been available for some time in Mumbai but service has stopped recently. The detection of gene defects in DYSF still remains a bottleneck since no Indian laboratory has ever embarked on this lengthy, time-consuming, costly task. Indian clinicians are therefore dependent on the goodwill of foreign molecular geneticists to confirm the diagnosis of their patients at the molecular level. Hopefully, an international initiative put forward and financially supported by the Jain Foundation ( www.jain-foundation.org ) enables the mutation detection of numerous patients worldwide. The Jain Foundation website gathers information on dysferlinopathy, patients and professionals can freely register in order to get more information and news, as well as to get prepared for further clinical trials.Therapeutic Perspectives Palliative treatment is so far the only available option in dysferlinopathy. Daily management, based on medical or surgical interventions, is largely supportive but definitely more efficient than any innovative therapy whatever its great appeal and its potential impact in the long run. A customized therapeutic approach is always preferable for each patient and must take into account the natural history of disease. Patients either with MM, LGMD 2B or DMAT have specific needs. The vast majority of patients with MM remain ambulatory throughout their life. Limb girdle muscular type 2B, conversely, often confines the patient to a wheelchair after two or three decades of disease progression, sometimes less. The DMAT patients require the use of callipers to oppose foot drop more efficiently. A multidisciplinary approach is always better and should combine the expertise of at least an adult neurologist, a rehabilitation specialist, and paramedics (occupational therapist, physiotherapist and social workers). It is of the utmost importance to follow up the patient regularly (once a year is a good tempo in most cases), even in the absence of curative medications. Not only to evaluate the disease progression and intervene accordingly but also to keep track of the patient in the perspective of clinical trials. Monitoring of cardio-respiratory functions is also something valuable even if such complications remain an exception in dysferlinopathies. Surgical procedures are rarely needed in dysferlin deficiencies. Surgical release of the Achilles tendons is sometimes useful in early stages of LGMD 2B in order to prolong ambulation but it is not a rule. In DMAT, ankle arhrodesis may also help in case of advanced foot drop with major functional impairment. Pharmacological medications have not proved to be of any benefit in dysferlinopathy. Anecdotal data suggest dantrolene could be beneficial to CK levels but without any impact at the clinical level. [68] The impact of corticosteroids is still subject to many discussions. Many patients misdiagnosed with PM erroneously received corticosteroids, sometimes for long duration, but none reported any dramatic clinical improvement. One randomized clinical trial is in progress in Germany to document this point in more detail. Other clinicians also work along the immune pathway to counteract the initiation of inflammation. Intravenous immunoglobulins (Iv-IG) and other immunosuppressive agents (some of them being more specific to certain lymphocyte populations) are also under consideration in therapeutic trials to come. Gene-based therapies are still far away from any clinical application in dysferlinopathy but they represent a great hope. Such therapeutic avenues are currently taking advantage from advances and technological breakthroughs made in other muscular dystrophies, especially in DMD. Besides, DMD and dysferlinopathy share many traits such as the critical size of the gene, the wide range of mutations and the comparable severity of the necrotizing process (even though DMD starts earlier and cardiomyopathy is not a part of the cardinal symptoms in dysferlinopathy). One good example of these promising techniques has recently been given by the Marseilles′ group which demonstrated that shortened versions of the DYSF gene could still be functional, a finding long established in DMD (the various so-called mini-dystrophins). This important discovery was based on the observation of a minimally affected 41-year-old patient with dysferlinopathy harboring a large DYSF deletion. The so-called mini-dysferlin derived from this patient has been able to rescue faulty processes of membrane repair in vitro. [69] Many other therapeutic avenues, such as exon-skipping, viral gene therapy and stem cell therapies are also being investigated. References
Copyright 2008 - Neurology India The following images related to this document are available:Photo images[ni08079f2.jpg] [ni08079f1.jpg] |
| |||||||||

{kind=link}
{kind=link}