 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
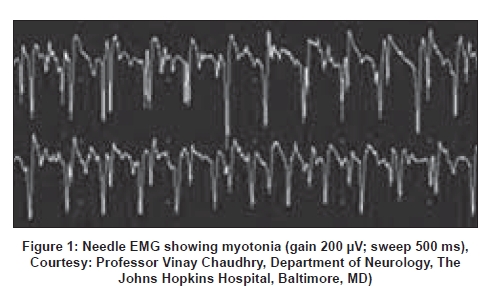
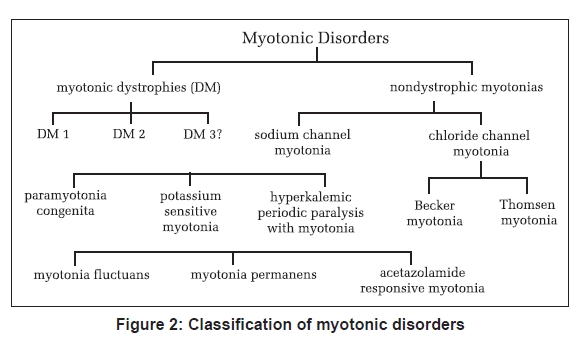
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 298-304 Review Article Myotonic disorders Mankodi Ami Department of Neurology, Johns Hopkins University, Baltimore, MD Date of Acceptance: 17-Aug-2008 Code Number: ni08080 Abstract Myotonia reflects a state of muscle fiber hyperexcitability. Impaired transmembrane conductance of either chloride or sodium ions results in myotonia. Myotonic disorders include the myotonic dystrophies and nondystrophic myotonias. Mutations in the genes encoding chloride (ClC-1) or sodium (SCN4A) channels expressed exclusively in skeletal muscle cause nondystrophic myotonias. Genetic defects in the myotonic dystrophies do not involve ion channel or its regulator proteins. Recent research supports a novel RNA-mediated disease mechanism of myotonia in the myotonic dystrophies. Myotonic dystrophy Type 1 is caused by CTG repeat expansion in the 3' untranslated region in the Dystrophia Myotonica Protein Kinase (DMPK) gene. Myotonic dystrophy Type 2 is caused by CCTG repeat expansion in the first intron in Zinc Finger Protein 9 (ZNF9) gene. The expanded repeat is transcribed in RNA and forms discrete inclusions in nucleus in both types of myotonic dystrophies. Mutant RNA sequesters MBNL1, a splice regulator protein and depletes MBNL1 from the nucleoplasm. Loss of MBNL1 results in altered splicing of ClC-1 mRNA. Altered splice products do not encode functional ClC-1 protein. Subsequent loss of chloride conductance in muscle membrane causes myotonia in the myotonic dystrophies. The purpose of this review is to discuss the clinical presentation, recent advances in understanding the disease mechanism with particular emphasis on myotonic dystrophies and potential therapy options in myotonic disorders.Keywords: Alternative splicing, chloride channel myotonia, myotonia, myotonia congenita, myotonic dystrophy, nondystrophic myotonia, nuclear inclusions, sodium channel myotonia Myotonia is the delayed relaxation of skeletal muscle fibers after voluntary muscle contraction. [1] Patients with myotonia may report painless muscle stiffness immediately upon initiating muscle activity after a period of rest. For example, inability to release hand grip after a strong handshake or trouble climbing stairs after a period of sitting. On physical examination, action myotonia can be elicited by isotonic muscle contraction such as asking the patient to make a tight fist or to grip and release the examiner′s fingers. Patients may have a lag in opening the eyes after initial tight eyelid closure. Myotonia improves with muscle exercise or repeated efforts, the so-called "warm-up phenomenon". Percussion myotonia is a prolonged muscle contraction after mechanical compression of the muscle with a reflex hammer. Percussion of the thenar eminence results in prolonged adduction of the thumb, and percussion on the extensor digitorum communis in the forearm while the wrist is hanging down results in prolonged extension of the wrist. Clinical myotonia is the cumulative result of electrical hyperexcitability of individual muscle fibers. Needle Electromyography (EMG) reveals spontaneous runs of motor unit potentials with a characteristic waxing and waning frequency and amplitude [Figure 1]. The frequency may range from 20 to 150 mHz with amplitudes of the myotonic potentials in the 10 to 1000 mV range. Myotonic discharges produce an unforgettable unique sound similar to a motorcycle motor or World War II dive bomber or a chainsaw. By definition, myotonic discharges last 500 ms or longer and should be identified in at least three areas of an individual muscle outside of an endplate region. [2] Persistent depolarization of the myotonic muscle is due to abnormal expression of ion channels in the muscle membrane. Point mutations or deletions in the genes encoding either the sodium channel ( SCN4A ) or chloride channel ( CLCN1 ) expressed in skeletal muscle result in myotonia without muscle atrophy or degeneration, the so-called nondystrophic myotonias. [3],[4] By contrast, DNA repeat expansion mutations causing myotonic dystrophies (DM) do not involve the ion channel or its regulatory proteins. Recent research has begun to unravel novel mechanisms of myotonia in DM. [5],[6],[7],[8] This review will focus on the current understanding of the disease mechanisms and treatment in DM and the nondystrophic myotonias (see classification of myotonic disorders in Figure 2). The Myotonic Dystrophies DM is dominantly inherited with a 50% risk of transmission from the affected parent to each child. Currently, two distinct mutations are known that lead to the clinical syndrome of DM. Myotonic dystrophy Type 1 (DM1) is caused by expansion of CTG repeats within 3′ untranslated region in the DMPK gene on Chromosome 19. [9] Myotonic dystrophy Type 2 (DM2) is caused by expansion of CCTG repeats within the first intron in the ZNF9 gene on Chromosome 3. [10] Most patients with DM phenotype have DM1 mutation which has an estimated prevalence of 1 in 8000 adult US population. Prevalence of DM2 varies with the population in different continents. For example, DM2 is as prevalent as DM1 in the studied population in Finland and Germany. [11] It is believed that DM mutations are not as prevalent in the Indian subcontinent; however, well-designed genetic epidemiological surveys are needed to better understand the disease prevalence in India. A search is on for new mutations given that some families with DM-like phenotype do not have either DM1 or DM2 mutations. The DNA repeat expansion remains stable throughout embryonic development. The repeats significantly vary in size in different tissues in an individual with significantly larger repeat sizes in muscle, heart and brain, the tissues most severely affected in DM. [12] The repeat size variability may increase with age. [13] There appears to be a rough correlation between the size of the repeat and the age of onset of clinical features for CTG < 400; [14] such correlation is lost for larger repeats. DM2 mutation is highly unstable with greater variations and much larger repeat sizes. Fortunately, the phenotype is not worse in DM2 patients with massive repeat expansion. In the same pedigree, family members can show a broad range of clinical severity and sizes of the repeats. Genetic anticipation refers to a more severe and earlier onset of phenotype in the subsequent generation secondary to a much larger increase in the repeat size. One such example is the congenital form of DM1 in which the size of CTG repeat increases from a few hundred base pairs to several thousand base pairs in the ovum and is transmitted from a mother to a child. There is a limit to the size of repeat expansion transmitted by sperm for unclear reasons. DM is the most common myotonic disorder in adults. Myotonia can be a presenting symptom but generally does not result in disability. Clinical diagnosis of DM can be easily made by the combination of myotonia and the characteristic pattern of muscle wasting and weakness. Myotonia is evident by delayed relaxation after a forceful sustained hand grip and percussion on thenar and wrist extensor muscles. Patients have prominent wasting and weakness of superficial facial muscles, levator palpebrae superioris, temporalis, masseter and palate with resultant ptosis, dysarthria and myopathic face. Swallowing may be impaired early in the disease. Sternocleidomastoids and distal muscles in the extremities are preferentially involved. Diaphragm, intercostal muscles, intrinsic muscles in hands, quadriceps, and external ocular muscles are often involved. Gastrocnemius, soleus, hamstrings, and pelvic girdle muscles are usually spared. Many patients with DM2 mutation present with prominent proximal muscle weakness, the so-called proximal myotonic myopathy. [15] Distal pattern of muscle weakness similar to DM1 can also be seen in patients with DM2. Cardiac involvement in the form of conduction delay, atrial flutter and fibrillation are common in both types of DM. Sudden cardiac death presumably related to arrhythmia is a leading cause of mortality in DM second only to respiratory complications. [16] Premature cataract before the age of 40 years is another common feature between the two diseases. Cataract can be present without skeletal muscle or cardiac disease and may lead to a new diagnosis of DM in a pedigree. Early cataract in DM has a characteristic Christmas tree-like appearance with multicolored refractile opacities visible in both anterior and posterior subcapsular regions in the lens when examined with a slit-lamp. Central nervous system disease characterized by apathy, somnolence, and inertia leads to psychosocial and occupational limitations jeopardizing the quality of life. Myotonic dystrophy patients frequently do not report symptoms and their functional abilities are deteriorated earlier and out of proportion to the extent of muscle weakness. Other systemic manifestations common to both types of DM include frontal balding, testicular atrophy, hypogammaglobulinemia, and insulin resistance. Difficulty with protecting airway and hypoventilation are common complications of anesthesia in DM1 patients. Respiratory and bulbar muscles are not severely affected in DM2. Muscle maldevelopment, hypotonia and mental retardation are prominent in congenital DM. Myotonia is not apparent until after the infancy. Poor fetal movement and polyhydramnios indicate that symptoms may begin as early as in intrauterine life. Severe weakness of face, jaw and elevated diaphragm with thin ribs on a chest X-ray may provide a clue to the diagnosis. Congenital DM is almost always due to large unstable CTG repeat expansions occurring during maternal transmission. Examination of the mother with milder clinical features of DM confirms the diagnosis. Congenital DM has never been reported in DM2. The DNA test is well established and is commercially available. Greater sensitivity and specificity and easy availability of the DM gene test has virtually eliminated the need for invasive muscle biopsy to establish the diagnosis. Muscle histology reveals prominent pyknotic nuclear clumps, increased internal nuclei, variable muscle fiber size, muscle fiber atrophy involving Type I fibers in DM1 and Type II fibers in DM2 and conspicuous absence of fibrosis, necrosis and regeneration. DM1 is caused by expansion of CTG repeat in the DMPK gene. [9] DM2 is caused by expansion of CCTG repeat in the ZNF9 gene. [10] DMPK, a serine threonine kinase shows no functional similarity to ZNF9 , a 7 zinc finger protein thought to bind RNA. Yet, both mutations result in quite similar clinical features including myotonia, myopathy, cardiac conduction defects, arrhythmia, cataracts and insulin resistance. In both diseases, the repeat transcribed in the mutant RNA forms inclusions in the nucleus. [17],[10] This finding suggested that the mutant RNA might have a significant role in the disease process. Mice that express expanded CUG repeats in skeletal muscle develop characteristic phenotype of myotonia and progressive myopathy. [18] Like in human DM, the mutant RNA containing expanded repeats form inclusions in the nuclei. Myotonia is an early and prominent symptom. Clinically, the CUG repeat mice develop muscle stiffness pronounced in hind limbs and lumbar paraspinals. The EMG shows true myotonic discharges in many muscles. Intracellular recordings in individual muscle fibers revealed markedly reduced (>90%) chloride conductance. [19] Expression analysis of ClC-1, a major chloride channel in skeletal muscle, showed altered splicing of ClC-1 mRNA. Abnormal splice products of ClC-1 do not encode functional ClC-1 protein and cause loss of chloride conductance that leads to myotonia. [19] Similar splicing abnormalities in ClC-1 mRNA and loss of chloride channel protein in muscle membrane are also seen in human DM1 and DM2. [19],[20] A large body of evidence indicates that the mutant DM RNA interferes with the activities of two protein families that act antagonistically to each other. Amongst many functions, these two groups of proteins directly act on RNA splicing. Whereas MBNL1 a protein in the muscleblind family promotes transition of splicing from fetal to adult exons, CUG binding protein or CUGBP1 helps retain the fetal exons. [21] CUGBP1, contrary to its name, binds to single-stranded RNA adjacent to repeats [22] and does not localize to the nuclear RNA foci in DM. [23] By unknown mechanisms, amounts and activity of this protein are increased in DM1 cells. [24] The amounts of CUGBP1 are not changed in skeletal muscle in DM2. [25] By contrast to CUGBP1, MBNL1 directly binds to the repeat rich double-stranded RNA hairpin [26] and localizes to the nuclear RNA foci in both types of DM. [23] A semiquantitative analysis showed more than 80% depletion of MBNL1 in nucleoplasm in DM1 and DM2 compared to normal controls. [25] The net result therefore is expression of fetal splice isoforms in adult tissues in DM. [21],[26] Does sequestration of MBNL protein(s) by repeat expansion RNA lead to loss of its function? Are MBNL proteins essential for processing of ClC-1 pre-RNA in myonucleus? If so, can loss of MBNL protein(s) by itself cause myotonia independent of the repeat expansion RNA? To address these questions, transgenic mice were generated with a specific defect in Mbnl gene so that these mice did not express the CUG repeat RNA binding isoforms of MBNL protein. [27] These mice developed myotonia, myopathy and cataracts reminiscent of human DM. In addition, these mice had processing defects of ClC-1 pre-mRNA in myonucleus and loss of ClC-1 protein in the muscle membrane similar to that observed in DM muscle. This study provides key evidence that MBNL proteins are at least one of the culprits that mediate toxic effects of DM RNA. To prove the point that loss of MBNL1 is primary mechanism for myotonia, a specific isoform of MBNL1 protein which is severely depleted in DM muscle was overexpressed in the skeletal muscle in the CUG repeat mice. Overexpression of MBNL1 restored splicing in ClC-1 resulting in reversal of ClC-1 channels expression in muscle corresponding with improvement in myotonia. [28] Nuclear RNA foci are seen in skeletal muscle, heart and brain, the most commonly and severely affected tissues in DM. [23],[29],[30] Depletion of MBNL1 has been demonstrated in all the three by immunofluorescence. [25],[29],[30] Developmentally regulated splicing of a broad range of target mRNAs is affected in all the three tissues. [25],[29],[30] (reviewed in [6] ) In summary, expanded repeats are transcribed in RNA and form discrete inclusions in nucleus. Mutant RNA sequesters MBNL1 and depletes MBNL1 from nucleoplasm. Loss of MBNL1 alone is sufficient to result in expression of fetal splice isoforms inappropriately in adult tissues. In addition to the ClC-1, several target pre-mRNAs have been identified that may have relevance in disease mechanism in DM (reviewed in [6] ). It is not clear whether the cumulative result of altered splice products in different tissues can entirely explain the complex phenotype in DM. For further reading on recent advances in the RNA pathogenesis in DM, please refer to the recently published excellent review articles. [5],[6],[7],[8] Insights into the disease mechanism have already paved the way for potential therapies which may become available in the near future to DM patients. For example, simple engineering of a splice site in ClC-1 mRNA such that it is no longer amenable to the altered splicing effect of CUG repeat RNA resulted in improvement of myotonia in CUG repeat mice. [31] Alternatively, overexpression of MBNL1 resulted in reversal of myotonia in CUG repeat mice. [28] However, myotonia is rarely disabling in DM. Patients often do not require specific medical therapy for myotonia. Management of DM remains at best supportive comprehensive care including patient education, genetic counseling, regular monitoring of cardiac rhythm, early detection of respiratory and swallowing problems, avoidance of anesthesia-related complications, cataract surgery, and provision of physical therapy, occupational therapy, speech and swallowing therapy, and community outreach social welfare programs. The Nondystrophic Myotonias The nondystrophic myotonias are pure skeletal muscle diseases without the involvement of the heart, brain, eye or other tissues. Myotonia can be dramatic and sometimes disabling. Emotional surprises, cold, potassium or exercise are potential triggers for myotonia. Muscle weakness and wasting are not prominent. The nondystrophic myotonias are ion channel disorders caused by conventional point mutations or deletions in the chloride or sodium channel genes with exclusive expression in skeletal muscle. [32],[33],[34],[35] Diagnosis is mostly based on clinical presentation by careful history and physical examination. Ancillary tests such as EMG and nerve conduction studies may be helpful. Gene tests are usually not required to make the diagnosis. Myotonia and other symptoms are easily manageable by activity modifications, avoiding certain triggers, and pharmacological therapies. Prognosis is generally excellent. Please refer to these excellent review articles for further reading. [3],[4]Chloride Channel Myotonia The nondystrophic myotonias due to chloride channelopathy include Thomsen′s myotonia congenita, Becker myotonia congenita, myotonia levior and fluctuating myotonia congenita. All forms of myotonia congenita are caused by mutations that result in loss of function of the chloride channel ClC-1, which is expressed exclusively in skeletal muscle membrane. [36] Potassium accumulation in transverse tubular system with each muscle action potential in the setting of low surface chloride conductance in skeletal muscle fibers result in myotonia. [37] More than 70 mutations in the CLCN1 gene have been identified in patients with myotonia congenita. [38] Mutations at the same gene locus can cause either autosomal dominant or recessive disease. Molecular testing for CLCN1 mutations is commercially available. The DNA testing is generally not indicated as it is very expensive and often there is a technical limitation to screen for so many mutations described in each of the myotonic disorders. Thomsen′s myotonia congenita Myotonia Levior and fluctuating myotonia congenita are likely clinical variations of Thomsen′s myotonia. Muscle hypertrophy is absent. Symptoms are milder and fluctuate with time. Becker′s myotonia congenita Sodium Channel Myotonia Myotonias secondary to sodium channelopathy are characterized by sensitivity to cold exposure, potassium ingestion and paradoxical worsening after muscle exercise in contrast to the warm-up phenomenon seen in the chloride channel myotonias. [36] All sodium channel myotonias are inherited in autosomal dominant pattern. Sodium channel myotonias are classified as paramyotonia congenita, potassium-sensitive myotonias which include myotonia fluctuans, myotonia permanens and acetazolamide-responsive myotonia, and hyperkalemic periodic paralysis with myotonia. [36] Like chloride channel myotonias, several different mutations are identified in pedigrees. [36] DNA testing for mutations is not indicated for it is expensive and there is obvious limitation to screen for so many mutations causing an individual disorder. There is no easy explanation for disease mechanism in each sodium channel myotonic disorder. It is believed that almost all mutations alter activation kinetics of the sodium channel and enhance prolonged sodium entry into muscle cells contributing to persistent depolarization, a physiological state of myotonia. [36] Care must be taken in each of these disorders to prevent rigidity and rhabdomyolysis during or immediately after surgery. Paramyotonia congenita The potassium aggravated myotonia Myotonia permanens is a rare and severe form of nondystrophic myotonia. Clinical presentation includes onset before age 10 years, severe generalized myotonia, and muscle hypertrophy. Muscle weakness is not prominent. Severe myotonia involving intercostal muscles may result in respiratory compromise with hypoxemia and acidosis. Potassium ingestion and exercise are the usual triggers for myotonia. The EMG reveals generalized myotonia with normal motor unit potentials. Mexiletine or Tocainide may provide partial relief from myotonic stiffness. Acetazolamide may help relieve exercise-induced muscle stiffness or cramps. Acetazolamide-responsive myotonia is characterized by generalized myotonia worsened by potassium ingestion, cold and fasting, and excellent recovery with acetazolamide. Patients present during childhood with progressive generalized myotonia which is easily evident on clinical examination and by EMG. Eyelid paramyotonia may be seen in some patients. Myotonic stiffness can be painful. Exercise generally has no significant effect on myotonia. Treatment includes acetazolamide with starting dose 125 mg daily with gradual titration up to 250 mg three times a day if required. Side-effects include kidney stone formation, parasthesia, nausea, confusion, mood irritability, depression, rash and liver function abnormalities. Regular monitoring of complete blood count and liver functions is recommended. Mexiletine can also relieve myotonia. Hyperkalemic periodic paralysis with myotonia Differential Diagnoses for Electrical Myotonia Electrical myotonia with variable muscle weakness can be seen in acid maltase deficiency, myotubular congenital myopathy, polymyositis, hypothyroidism, malignant hyperthermia and exposure to certain drugs including clofibrate, HMG CoA inhibitors, propranolol, cyclosporine, colchicine, and penicillamine. Myotonia-like discharges can be seen in Schwartz Jampel syndrome and Isaac′s syndrome of neuromyotonia. Continuous muscle fiber activity in these disorders does not wax and wane like true myotonia. These disorders can also be easily diagnosed based on associated clinical features and distinct gene mutations. In summary, myotonic disorders are easily recognized by careful history and physical examination. Expensive DNA tests are not routinely indicated. Myotonia results from abnormal expression of either chloride or sodium channels in skeletal muscle. Disease mechanism of myotonia in nondystrophic myotonias is straightforward from conventional mutations in the genes encoding either chloride or sodium channels. In DM, myotonia results from impaired transmembrane chloride conductance by an unprecedented RNA-mediated disease mechanism. Mutant DM RNA sequesters MBNL1 and depletes MBNL1 in nucleoplasm. Subsequent loss of MBNL1 function leads to altered splicing of ClC-1. Abnormal splice products do not encode functional ClC-1, the major chloride channel in skeletal muscle. Loss of ClC-1 protein in muscle membrane results in impaired chloride conductance and myotonia. Mutant DM RNA interferes with splicing of several other transcripts in the skeletal muscle, heart and brain. Further research is needed to determine if abnormalities in intracellular metabolism of select target RNAs, splicing or otherwise, can explain multisystem involvement in DM. Initial stages of understanding the disease mechanism has already opened a new era of novel therapies in DM. References
Copyright 2008 - Neurology India The following images related to this document are available:Photo images[ni08080f1.jpg] [ni08080f2.jpg] |
| |||||||||

{kind=link}
{kind=link}