
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 56, Num. 3, 2008, pp. 305-313
|
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 305-313
Review Article
Autoimmune disorders of the neuromuscular junction
Vincent Angela
Department of Clinical Neurology, Neurosciences Group, Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, Oxford OX3 9DS
Correspondence Address:Neurosciences Group, Department of Clinical Neurology, Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, Oxford OX3 9DS
angela.vincent@imm.ox.ac.uk
Date of Acceptance: 04-Sep-2008
Code Number: ni08081
Abstract
The neuromuscular junction (NMJ) is a specialized synapse with a complex structural and functional organization. It is a target for a variety of immunological disorders and these diseases usually respond well to immunotherapies. The understanding of the immunological basis of myasthenia gravis, the most common neuromuscular junction disorder, has improved in the recent years. Most patients have antibodies to the acetylcholine receptor (AChR), but around 10% have AChR antibodies that are only identified by novel methods, and up to 5% have muscle-specific kinase antibodies which define a different subgroup of myasthenia. The spectrum of antibodies and their pathophysiological aspects are being elucidated. Even though less common, Lambert Eaton myasthenic syndrome (LEMS) is important to recognize. The abnormality in LEMS is a presynaptic failure to release enough packets of ACh, caused by antibodies to the presynaptic voltage-gated calcium channels. More than half these patients have a small cell carcinoma of lung. Acquired neuromyotonia (NMT) is a condition associated with muscle hyperactivity. Clinical features include muscle stiffness, cramps, myokymia, pseudomyotonia and weakness. The immune mechanisms of acquired NMT relate to loss of voltage-gated potassium channel function. This review will focus on the important recent developments in the immune-mediated disorders of the NMJ.
Keywords: Gravis, Lambert Eaton myasthenic syndrome, neuromyotonia, neuromuscular junction, autoimmunity, acetylcholine receptor, voltage-gated channel
The Neuromuscular Junction
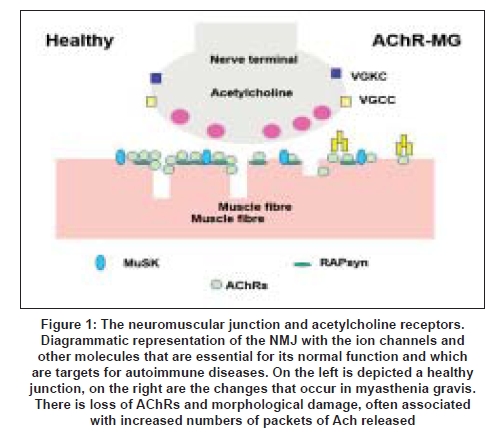
The neuromuscular junction (NMJ) is a prototypic synapse although its structure is rather different from those of the central nervous system (CNS). The unmyelinated motor nerve terminals are separated from the postsynaptic membrane by a cleft that contains a basal lamina. This includes many proteins such as collagens, laminins, fibronectin and perlecan which help anchor some of the key elements involved in NMJ development and function; for instance acetylcholine esterase (AChE) is localized via ColQ, a collagen-like molecule, to the basal lamina. Agrin and neuregulins, secreted from the nerve terminal, are bound by the basal lamina and interact with their receptors, playing an important role in the location of postsynaptic membrane proteins, voltage-gated calcium channels and the dystroglycans. The postsynaptic membrane at the NMJ forms a series of deep folds. The acetylcholine receptors (AChRs) are found at the top one-third of these folds, whereas the voltage-gated sodium channels are anchored at the bottom of the folds. The development of the NMJ is a fascinating area of research [1] and many of the proteins involved are relevant to disease (see Figure 1 for a simple representation).
The nerve action potential opens voltage-gated calcium channels (VGCCs) that are located in the motor nerve terminal . The resulting influx of calcium leads to the release of about 30 (in human muscle) individual packets of acetylcholine (ACh). Some of the ACh is hydrolysed by AChE but about 65% reaches the AChRs on the postsynaptic membrane. Binding of two ACh to each AChR leads to the opening of the AChR-associated ion channel, influx of cations (mainly sodium) and generation of an endplate potential (EPP). The EPP in normal human muscles is around 20-30 mV. Miniature EPPs (MEPPs) are the result of the spontaneous release of single packets of ACh and their amplitudes are much smaller (around 1 mV) and generally reflect the density of functional AChRs.
The EPP rapidly depolarises the postsynaptic membrane and, when this reaches a critical firing threshold, the voltatge-gated sodium channels open and an action potential is propagated along the muscle fiber leading to contraction. The extent to which the EPP exceeds that necessary to initiate the action potential is usually called the safety factor for neuromuscular transmission. [2] The EPP is short-lived because the AChRs close spontaneously, ACh dissociates and escapes by diffusion or is hydrolysed by AChE. The calcium channels also close spontaneously. Opening of voltage-gated potassium channels on the presynaptic membrane is important in restoring the membrane potential and limiting calcium channel opening.
The topic of this review is the chronic conditions caused by serum autoantibodies and which are treatable by immunotherapies. The NMJ is particularly vulnerable to circulating factors because it has no blood-brain barrier. This is well illustrated by envenomation by snakes, spiders, scorpions and other species, that results in NMJ paralysis or hyperexcitability. [3] Some of these neurotoxins are particularly valuable in studying the NMJ and for tagging the proteins for autoantibody assays, as will be described below.
Myasthenia Gravis
Clinical features
Myasthenia gravis (MG) usually presents in young adult or later adult life as muscle weakness and excessive fatigue during repetitive movements. It most often involves the extraocular muscles of the eye with double vision and ptosis at onset, but usually progresses to generalized weakness. Involvement of the facial and bulbar muscles is particularly disabling and respiratory involvement can be life-threatening. Cholinesterase inhibitors, by prolonging the action of ACh, tend to lead to clinical improvement. [4],[5] The history of myasthenia research is fascinating [6] but cannot be reviewed here. The essential findings in the 1960s and 1970s were that the MEPP amplitudes are substantially reduced in muscle biopsies from MG patients [7] and that this is due to loss of the postsynaptic AChRs. [8] The latter finding depended on the discovery of bungarotoin, a snake toxin that binds specifically to the AChRs. [9] At much the same time, purification of the AChR from electric organs of certain fish, and immunization of rabbits with the purified AChRs, led to the first experimental autoimmune myasthenia gravis (EAMG) model by Patrick and Lindstrom. [10] Lindstrom and his colleagues then detected antibodies to AChRs in the serum of MG patients, establishing the radioimmunoprecipitation assay that has changed little to this day. [11]
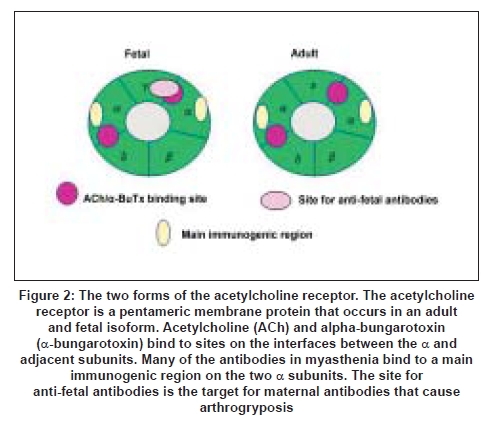
The adult and fetal AChRs are membrane proteins that differ in only one of the four subunits [Figure 2]. The levels of AChR antibodies vary widely between patients and there is only modest correlation with clinical severity. This is probably because the antibodies are highly heterogeneous in their characteristics and the epitopes to which they bind on the AChR. [12] However, during treatments such as plasma exchange the levels of AChR antibody correlated well with clinical severity within an individual. [13] This clinical experiment established the role of the antibodies in causing the disease. Equally important was the passive transfer of purified IgG from MG patients to mice that resulted in NMJ deficits with reduced AChRs . [14]
Clinical Heterogeneity
Ocular myasthenia gravis
It is unclear why the extraocular muscles are so often the first to be affected and may remain the only muscles involved. Indeed, they should be resistant to fatigue with high blood flow, mitochondria content and metabolic rate. [15] However, the motor unit sizes are small and the firing frequencies are high, and some of the muscle fibers have multiple NMJs in which the EPPs, rather than action potentials, are directly responsible for activating the contractile apparatus. This means that any reduction in the EPP could have a direct effect on the strength of muscle contraction. There has been some controversy about the role of fetal rather than adult AChRs at extraocular muscle endplates, [16] but another factor that might make them more susceptible is the low expression of complement regulators; [17] this would make them more vulnerable to complement-mediated damage. Of possible relevance to this, in the Lambert Eaton myasthenic syndrome (LEMS, see below), in which complement-mediated damage is not thought to occur, ocular muscle weakness is uncommon.
Generalized Acetylcholine Receptor
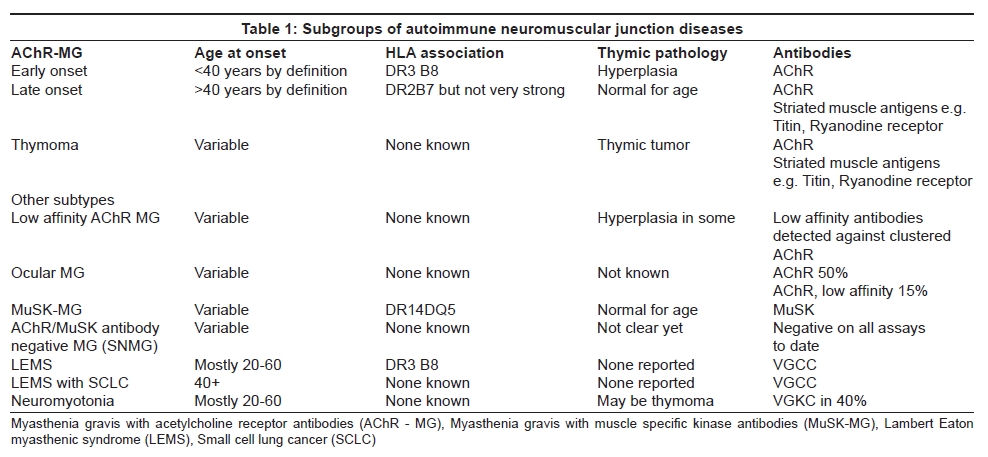
Most patients in the Western world progress to generalized weakness, although in some Eastern countries ocular MG, particularly in young children, is very common. [18] Based on age at onset, HLA associations, AChR antibody positivity and thymic pathology the patients can be divided into early onset, late onset and thymoma-MG . [19] [Table 1]
In the older patients, the Human leukocyte antigen (HLA) association is less marked and the thymus is essentially normal for age. Despite these important subgroups, the disease mechanisms appear to be very similar (see below). The late onset patients are being recognized much more frequently in the population and may well be underdiagnosed or misdiagnosed as stroke or motor neuron disease. [20]
Neonatal Myasthenia Gravis and Arthrogryposis Multiplex Congenita A relatively small proportion of babies born to MG mothers have neonatal MG, due to the placental transfer of the maternal antibodies. In a small number of reported cases, the mothers give birth to babies with arthrogryposis multiplex congenita. This condition, which involves multiple fixed joint contractures and other abnormalities, can be caused by anything that reduces fetal movement in utero. When it is associated with maternal AChR antibodies it typically recurs in subsequent pregnancies unless the mother is adequately treated. Since some mothers have little, or even no, evidence of MG themselves, multiple affected offspring have been reported in some cases. [21] This condition appears to be caused by maternal antibodies that inhibit the function of the fetal form of the AChR paralyzing the developing fetus. [22] We established a maternal-to-fetal passive transfer model of this condition [23] and the possibility of maternal antibodies should be considered in other developmental conditions that recur in each consecutive pregnancy.
The Cause of Myasthenia Gravis Despite many years of study, the cause(s) of MG are unknown. The antibodies are highly specific, generally high affinity for the AChR and variable in their characteristics. The binding sites on the AChR can be defined by competition with monoclonal antibodies raised against purified AChRs. [24],[25] Many bind to a "main immunogenic region" (MIR) on the two AChR alpha subunits, but many antibodies bind to the other subunits, including the fetal-specific gamma subunit. Overall, the characteristics suggest that the human AChR is the immunogen and that the antibodies do not arise as the result of a cross-reaction with microbial antigens, as was proposed in the 1980s. However, it is possible that a low-affinity or cross-reactive antibody may lead subsequently to a higher affinity reaction against the AChR. [26] We now believe that lower-affinity antibodies do exist in some patients. [27]
Mechanisms of Disease
The loss of AChRs at the NMJ is the underlying disease mechanism. This leads to reduced amplitude of the EPPs. The EPPs are often supra-threshold to begin with (and therefore induce an action potential in the muscle) but, during repetitive activity, they rapidly become sub-threshold leading to muscle weakness. The loss of AChRs results from several different processes. Relatively few antibodies directly block the function of the ACh-induced ion channel, but these antibodies can be important in some patients [28],[29] and some MG sera appear to have a transient inhibitory effect on AChR function in vitro. [30] A more important mechanism is a reduction in AChRs due to the cross-linking of AChRs by divalent antibodies. [31] This results in a temperature-dependent loss of AChR which can best be demonstrated in muscle cell lines in cultures, but has also been demonstrated at the mouse NMJ. [32],[33] Interestingly, there was an increase in synthesis rate of new AChRs [34] in these experiments and in MG muscle biopsies, there is an increase in expression of the AChR. [35]
Probably the most important mechanism of AChR loss is complement-mediated destruction of the postsynaptic membrane. [36] The AChR antibodies are usually IgG1 or IgG which are both complement-activating subclasses. IgG [37] and complement components C3 and C9, and of the membrane attack complex (MAC), are localized at the NMJ [38] and AChR numbers appear to be inversely related to the amount of complement. There is also morphological damage to the endplates and simplification of the postsynaptic folds [Figure 1]. This could be important as the voltage-gated sodium channels responsible for initiating the action potential are located at the bottom of the folds. [39]
Several factors could modify the degree of AChR loss and thereby disease severity. Complement regulatory proteins are present which probably reduce the damage caused by complement-activating antibodies [40] and as mentioned above there is increased AChR synthesis. In addition, the number of ACh packets released is increased at individual endplates. [41] All of these factors are likely to be genetically regulated and will modify the severity of the disease and perhaps explain some of the variability between individuals, within an individual in different muscles and at different times.
The Thymus in Myasthenia Gravis
There are two main thymic pathologies in MG patients [Table 1]. Early-onset MG is associated with enlarged thymus glands with frequent lymphocytic infiltrates and germinal centers, similar to those found in lymph nodes. These glands, unusually, contain B cells and plasma cells as well as T cells. Moreover, in the thymic medulla, and sometimes surrounding or within the germinal centers, there are muscle-like myoid cells that express fetal AChR [42] and other muscle proteins. [43] It is known that the thymic cells can actively produce AChR antibody when cultured in vitro [44] and removal of the thymus results in a moderate fall in AChR antibody although the levels usually plateau out after some months; [45] nowadays most patients are also given immunosuppressive treatments. Further work on the thymic pathology has also shown that complement is activated on the abnormal epithelium, and on the myoid cells; it seems very likely that the AChRs expressed by these myoid cells initiate or maintain the immune response in early-onset MG. [46] A recent report describes some of the factors that control AChR expression in these cells. [47]
About 10% of MG patients have a thymoma, usually presenting in middle age and sometimes associated with other autoimmune disorders. Thymomas are epithelial tumors but often contain many T lymphocytes (and few B cells). They appear to be capable of exporting mature T cells [48] but these cells may lack the normal regulatory mechanisms. [49] The role of the thymoma is somewhat different from that of the thymus in early-onset MG. The AChR is not generally found in the thymoma itself, although there may be AChR subunits expressed individually in different epithelial cells. However, if AChR-sensitive T cells escape from the disordered thymic environment, they could get stimulated by the presence of AChRs on the myoid cells present in the adjacent "normal" thymus. Some of these possibilities are discussed by Vincent et al. [26] Thymectomy generally does not lead to clinical improvement and most patients require immunosuppressive treatments.
The thymus in late-onset MG is usually considered to be atrophic or "normal for age". However, there are few histological studies on this age group and thymectomy is seldom performed for MG patients over the age of 60 years, unless a thymoma is present.
Myasthenia Gravis without Acetylcholine Receptor Antibodies Up to 20% of patients with generalized MG do not have detectable AChR antibodies by current laboratory methods, but their disease is also antibody mediated because they respond to plasma exchange, and their IgG can passively transfer disease to mice. [50] A number of studies over many years have tried to define the nature of the antibodies in these patients.
Myasthenia Gravis with Muscle-specific Kinase Antibodies
A proportion of patients without AChR antibodies have instead antibodies to a protein called MuSK (muscle-specific kinase), which is restricted to the NMJ in adult muscle. MuSK is essential for the development of the NMJ [51] but its role in adult muscle is less clear although it probably plays an important role in maintaining the structure of the postsynaptic membrane. [52] MuSK-MG often presents with ocular involvement, but frequently evolves to include severe bulbar and facial weakness, sometimes with marked muscle atrophy in these muscles. [53],[54]
Antibodies to MuSK [55] were demonstrated in around 70% of patients without AChR antibodies, and in none of the AChR antibody positive patients. Subsequently radioimmunoprecipitation assays found the antibodies in varying proportions of patients and the incidence seems to vary throughout the northern hemisphere (there are insufficient data from the southern hemisphere). [56]
There are several interesting differences between MuSK-MG and typical AChR-MG patients. Firstly, the thymus in MuSK-MG is usually normal with very small and infrequent germinal centers [57] and little complement activation. [46] Secondly, the disease is associated with HLA DR14-DQ5 [58] suggesting that the genetic susceptibility is very different from typical MG [Table 1]. Moreover, the MuSK antibodies are mainly IgG4 [59] unlike the IgG1 and IgG3 of AChR antibodies. IgG4 is not a strong activator of complement and complement deposition was not found at MuSK NMJs in one study of limb muscle biopsies. [60] Curiously, there was little evidence of AChR loss either, and therefore the mechanisms by which MuSK-MG patients become weak are not clear.
There have not yet, however, been any detailed studies of neuromuscular transmission in the most affected muscles. The limb muscles are often relatively unaffected and limb muscle electrophysiology may be normal in MuSK-MG, [60] while facial muscles are abnormal; [61],[62] this suggesting that the MuSK antibodies are particularly pathogenic in these muscles, as supported by magnetic resonance imaging (MRI) studies. [54] Further in vitro and animal studies are required to determine the role of MuSK in adult muscle, and to look for differences between facial and limb NMJs.
Myasthenia without Muscle-specific Kinase or Acetylcholine Receptor Antibodies
There remain approximately 10% of MG patients who have neither MuSK nor AChR antibodies by the radioimmunoprecipitation methods used worldwide. However, we observed that these patients have clinical symptoms indistinguishable from those of typical AChR-MG patients, although they are often less severely affected. Moreover, their thymus glands show hyperplasia and germinal centers very similar to those of AChR-MG patients. [46],[57] Their sera frequently inhibited AChR function in vitro and seemed to increase desensitization of the AChR, but this effect appeared to be reversed by washing, [30],[63] suggesting that perhaps these antibodies were low affinity rather than the high affinity antibodies detected in serum.
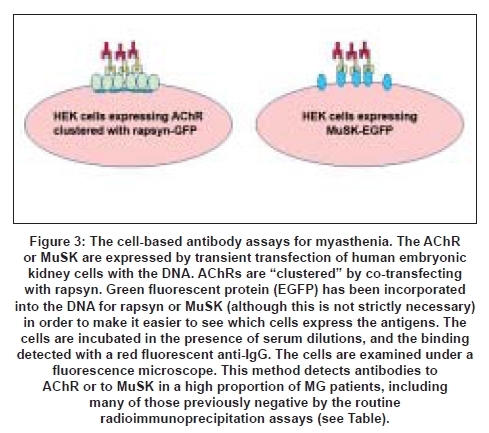
We hypothesized that these patients had antibodies that bound to the AChR with low affinity in solution but would be able to bind to the AChRs when they were expressed at the NMJ where the density of the receptors is very high. To reproduce this high density, we expressed AChRs in a cell line and clustered the receptors with the clustering protein rapsyn [Figure 1]. We were then able to detect the presence of antibodies in about 60% of the sera that were negative for binding to AChR in solution. [27] It appears that this "cell-based" method [Figure 3] can also be applied to measuring antibodies to MuSK, and should provide a more sensitive assay for all MG patients. [27]
The Lambert Eaton Myasthenic Syndrome
Clinical features
The Lambert Eaton myasthenic syndrome is another autoimmune form of NMJ disorder, but it is also a prototype paraneoplastic neurological syndrome as about 50% of the patients have small cell lung cancer. The distribution of muscle weakness is different from MG as the trunk and legs are most often involved, and ocular involvement is uncommon. Autonomic symptoms (dry mouth, constipation, impotence) are frequent. [64] Typically, weakness improves during sustained effort as can be shown clinically or by electromyography. The compound muscle action potential is very small but increases during high-frequency stimulation or following a brief period of voluntary contraction. The reflexes are absent or depressed but may become stronger after voluntary contraction.
Immunological basis
In patients with small cell lung cancer (SCLC) it is highly likely that the immune response is primarily directed against the tumor (see below), but in the remaining patients, who have a chronic disease, the cause is unknown. An HLA association with HLA B8DR3 suggests an underlying genetic predisposition to autoimmunity [65] and these patients often have other autoimmune disorders, such as thyroid disease or diabetes. [64] These observations led to the first experimental studies implicating the immune system. It was shown that patients with LEMS responded clinically to plasma exchange, and daily injection of LEMS IgG antibodies into mice reproduced the principal neurophysiologic changes of LEMS. [66]
Mechanisms of disease
The defect in LEMS is a presynaptic failure to release enough packets of ACh. The EPPS are very small, as they are in MG, but the MEPPS are of normal amplitude. [67] In these classic studies, repetitive stimulation of the nerve showed that the endplate potential increased during repetitive stimulation and also when extracellular calcium was raised. These results suggested a possible defect in the presynaptic voltage-gated calcium channels (VGCC).
In the 1980s Engel and colleagues used freeze fracture electron microscopy to study the motor nerve terminals in LEMS and in a passive transfer model of the disease (see below). They identified double parallel rows of intramembranous particles in the motor nerve terminal membrane which are thought to represent VGCCs. These channels were markedly reduced in number and their distribution was very abnormal. [68]
Mice injected with LEMS IgG showed similar changes to those described in the patients. [69] Moreover, IgG could be demonstrated on the motor nerve terminal, apparently at the position where VGCCs are thought to be concentrated. [70] Other evidence implicated divalent binding of antibodies leading to reduction in the VGCCs, and complement did not seem to be involved. [71]
It was some time before the presence of antibodies to the VGCCs was unequivocally demonstrated. VGCCs are a family of transmembrane proteins that can be distinguished by various drugs and neurotoxins. w-Conotoxin (w-CmTx) is a snail toxin that binds to the P/Q-type VGCC and inhibits neuromuscular transmission. [72] Antibodies in LEMS can be detected by radioimmunoprecipitation of VGCCs extracted from human or mammalian cerebellum that are labeled with 125 1- w-Conotoxin (w-CmTx) MVIIC. [73],[74] The antibodies are present in over 85% of patients and are highly specific for the disease, although they can be found in some patients with SCLC without LEMS. [75] SCLCs express VGCCs on their surface, and LEMS IgG reduces the numbers of VGCC in these cells and in other cell lines expressing the calcium channels. [76] Just as AChR synthesis is increased in MG muscle, so also there is likely to be increased synthesis of P/Q-type VGCC or alternative channels in motor nerves in LEMS. [77] P/Q-type VGCCs are also present at autonomic synapses explaining the frequent autonomic involvement in these patients. [78]
Acquired Neuromyotonia
Clinical features
Acquired neuromyotonia (NMT) is a condition associated with muscle hyperactivity and probably under-diagnosed as it is not life-threatening. Clinical features include muscle stiffness, cramps, myokymia, pseudomyotonia and weakness. These are most common in the limbs and trunk but can be found in isolated muscle groups. The muscle hyperactivity continues during sleep and excessive sweating is a common complaint. The typical finding on electromyography is spontaneous motor unit discharges occurring in distinctive doublets, triplets, or longer runs with high intraburst frequency. [79] Most of this activity is generated distally, perhaps at the NMJ itself, but in some cases more proximally. Cramp fasciculation syndrome may form part of the same clinical spectrum. [80]
Immunological basis
As with LEMS, the first clinical observations pointed to an autoimmune basis. Although no HLA association has yet been demonstrated, other autoimmune diseases are commonly present, and about 20% of patients have a thymoma which can predate the symptoms or be identified after presentation of the neurological disease. Some patients also have MG. [80] However, in contrast to MG and LEMS, some patients appear to have a monophasic illness that recovers spontaneously within a few months or years. There is circumstantial evidence that these forms of the disease occur following an infection or other immune response; one patient developed the disease after multiple wasp stings. [81]
Immune mechanisms
Although neuromyotonia is a peripheral disease, the cerebrospinal fluid may contain oligoclonal bands, and since it can be associated with other autoimmune diseases and thymoma, plasma exchange and passive transfer experiments were undertaken. [82] Injection of IgG into mice resulted in a modest increase in the number of packets of ACh released, and IgG also increased neuronal activity in dorsal root ganglion cultures. [83] Importantly, these findings were very similar to those in the presence of low concentrations of potassium channel blockers such as 4-aminopyridine. This suggested that loss of voltage-gated potassium channel (VGKC) function may underlie the electrophysiological findings in patients.
VGKCs are a large family of potassium channels, and the full spectrum of VGKC involved in neuromyotonia has not been explored. The antibodies are currently measured by radioimmunoprecipitation of VGKC Kv1.1, 1.2 and 1.6 that are conveniently identified by binding of the snake toxin, dendrotoxin. [84] Kv1.1 and 1.2 are highly expressed in the peripheral nervous system, particularly in the juxtaparanodal region of the nodes of Ranvier [85] and this is the most likely target for the antibodies, but they may also be expressed at the motor nerve terminal.
Antibodies to VGKCs can also be detected by immunostaining of xenopus oocytes or cell lines engineered to express the different Kv subtypes individually. [84],[86] The precise specificity of the antibodies is difficult to determine as the VGKCs are tetrameric proteins made up of different combinations of different Kv subunits. There are several reports that the antibodies, or IgG fractions, reduce potassium currents in cells transfected with the Kv1.2 subtype [87] and that this does not require complement. [88] It is likely that the antibodies act by reducing the VGKC numbers and that complement-mediated attack on the motor nerves does not occur. [89]
Central Nervous System Disease with Voltage-gated Potassium channel Antibodies Although the typical neuromyotonia patient does not have central symptoms, some have sensory nerve involvement and sleep problems or anxiety are not infrequent. [80] Some patients have severe neuromyotonia with additional autonomic and marked central symptoms. This is usually called Morvan′s syndrome. [90] In addition, patients with subacute onset of limbic encephalitis, with memory loss, seizures and disorientation [20] or epilepsy with minimal cognitive involvement, [91] are increasingly being recognized. It is still unclear as to which antibody specificity determines the clinical phenotype or whether other factors are involved.
References
| 1. | Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2001;2:791-805. Back to cited text no. 1 [PUBMED] [FULLTEXT] |
| 2. | Wood SJ, Slater CR. Safety factor at the neuromuscular junction. Prog Neurobiol 2001;64:393-429. Back to cited text no. 2 [PUBMED] [FULLTEXT] |
| 3. | Hodgson WC, Wickramaratna JC. In vitro neuromuscular activity of snake venoms. Clin Exp Pharmacol Physiol 2002;29:807-14. Back to cited text no. 3 [PUBMED] [FULLTEXT] |
| 4. | Drachman DB. Myasthenia gravis. N Engl J Med 1994;330:1797-810. Back to cited text no. 4 [PUBMED] [FULLTEXT] |
| 5. | Vincent A, Palace J, Hilton-Jones D. Myasthenia gravis. Lancet 2001;357:2122-8. Back to cited text no. 5 [PUBMED] [FULLTEXT] |
| 6. | Vincent A. Unravelling the pathogenesis of myasthenia gravis. Nat Rev Immunol 2002;2:797-804. Back to cited text no. 6 [PUBMED] [FULLTEXT] |
| 7. | Elmqvist D, Hofmann WW, Kugelberg J, Quastel DM. An electrophysiological investigation of neuromuscular transmission in myasthenia gravis. J Physiol 1964;174:417-34. Back to cited text no. 7 [PUBMED] [FULLTEXT] |
| 8. | Fambrough DM, Drachman DB, Satyamurti S. Neuromuscular junction in myasthenia gravis: Decreased acetylcholine receptors. Science 1973;182:293-5. Back to cited text no. 8 [PUBMED] [FULLTEXT] |
| 9. | Chu NS. Contribution of a snake venom toxin to myasthenia gravis: The discovery of alpha-bungarotoxin in Taiwan. J Hist Neurosci 2005;14:138-48. Back to cited text no. 9 [PUBMED] [FULLTEXT] |
| 10. | Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptor. Science 1973;180:871-2. Back to cited text no. 10 [PUBMED] [FULLTEXT] |
| 11. | Lindstrom JM, Seybold ME, Lennon VA, Whittingham S, Duane DD. Antibody to acetylcholine receptor in myasthenia gravis: Prevalence, clinical correlates and diagnostic value. Neurology 1976;26:1054-9. Back to cited text no. 11 [PUBMED] |
| 12. | Vincent A, Whiting PJ, Schluep M, Heidenreich F, Lang B, Roberts A, et al. Antibody heterogeneity and specificity in myasthenia gravis. Ann N Y Acad Sci 1987;505:106-20. Back to cited text no. 12 [PUBMED] [FULLTEXT] |
| 13. | Newsom-Davis J, Pinching AJ, Vincent A, Wilson SG. Function of circulating antibody to acetylcholine receptor in myasthenia gravis: Investigation by plasma exchange. Neurology 1978;28:266-72. Back to cited text no. 13 [PUBMED] |
| 14. | Toyka KV, Brachman DB, Pestronk A, Kao I. Myasthenia gravis: Passive transfer from man to mouse. Science 1975;190:397-9. Back to cited text no. 14 [PUBMED] [FULLTEXT] |
| 15. | Yu Wai Man CY, Chinnery PF, Griffiths PG. Extraocular muscles have fundamentally distinct properties that make them selectively vulnerable to certain disorders. Neuromuscul Disord 2005;15:17-23. Back to cited text no. 15 [PUBMED] [FULLTEXT] |
| 16. | MacLennan C, Beeson D, Buijs AM, Vincent A, Newsom-Davis J. Acetylcholine receptor expression in human extraocular muscles and their susceptibility to myasthenia gravis. Ann Neurol 1997;41:423-31. Back to cited text no. 16 [PUBMED] |
| 17. | Kaminski HJ, Li Z, Richmonds C, Lin F, Medof ME. Complement regulators in extraocular muscle and experimental autoimmune myasthenia gravis. Exp Neurol 2004;189:333-42. Back to cited text no. 17 [PUBMED] [FULLTEXT] |
| 18. | Zhang X, Yang M, Xu J, Lang B, Wang W, Vincent A. Clinical and serological study of myasthenia gravis in HuBei Province, China. J Neurol Neurosurg Psychiatry 2007;78:386-90. Back to cited text no. 18 [PUBMED] [FULLTEXT] |
| 19. | Compston DA, Vincent A, Newsom-Davis J, Batchelor JR. Clinical, pathological, HLA antigen and immunological evidence for disease heterogeneity in myasthenia gravis. Brain 1980;103:579-601. Back to cited text no. 19 [PUBMED] [FULLTEXT] |
| 20. | Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, et al. Potassium channel antibody-associated encephalopathy: A potentially immunotherapy-responsive form of limbic encephalitis. Brain 2004;127:701-12. Back to cited text no. 20 [PUBMED] [FULLTEXT] |
| 21. | Polizzi A, Huson, S M, Vincent A. Teratogen update: Maternal myasthenia gravis as a cause of congenital arthrogryposis. Teratology 2000;62:332-41. Back to cited text no. 21 |
| 22. | Vincent A, Newland C, Brueton L, Beeson D, Riemersma S, Huson SM, et al. Arthrogryposis multiplex congenita with maternal autoantibodies specific for a fetal antigen. Lancet 1995;346:24-5. Back to cited text no. 22 [PUBMED] |
| 23. | Jacobson L, Polizzi A, Morriss-Kay G, Vincent A. Plasma from human mothers of fetuses with severe arthrogryposis multiplex congenita causes deformities in mice. J Clin Invest 1999;103:1031-38. Back to cited text no. 23 [PUBMED] [FULLTEXT] |
| 24. | Tzartos SJ, Barkas T, Cung MT, Mamalaki A, Marraud M, Orlewski P, et al. Anatomy of the antigenic structure of a large membrane autoantigen, the muscle-type nicotinic acetylcholine receptor. Immunol Rev 1998;163:89-120. Back to cited text no. 24 [PUBMED] |
| 25. | Whiting PJ, Vincent A, Schluep M, Newsom-Davis J. Monoclonal antibodies that distinguish between normal and denervated human acetylcholine receptor. J Neuroimmunol 1986;11:223-35. Back to cited text no. 25 [PUBMED] |
| 26. | Vincent A, Willcox N, Hill M, Curnow J, MacLennan C, Beeson D. Determinant spreading and immune responses to acetylcholine receptors in myasthenia gravis. Immunol Rev 1998;164:157-68. Back to cited text no. 26 [PUBMED] |
| 27. | Leite MI, Jacob S, Viegas S, Cossins J, Clover L, Morgan BP, et al. IgG1 antibodies to acetylcholine receptors in "seronegative" MG. Brain;131:1940-52. Back to cited text no. 27 |
| 28. | Weinberg CB, Hall Z W. Antibodies from patients with myasthenia gravis recognize determinants unique to extrajunctional acetylcholine receptors. Proc Natl Acad Sci USA 1979;76:504-8. Back to cited text no. 28 |
| 29. | Burges J, Wray DW, Pizzighella S, Hall Z, Vincent A. A myasthenia gravis plasma immunoglobulin reduces miniature endplate potentials at human endplates in vitro. Muscle Nerve 1990;13:407-13. Back to cited text no. 29 [PUBMED] |
| 30. | Bufler J, Pitz R, Czep M, Wick M, Franke C. Purified IgG from seropositive and seronegative patients with mysasthenia gravis reversibly blocks currents through nicotinic acetylcholine receptor channels. Ann Neurol 1998;43:458-64. Back to cited text no. 30 [PUBMED] |
| 31. | Heinemann S, Bevan S, Kullberg R, Lindstrom J, Rice J. Modulation of acetylcholine receptor by antibody against the receptor. Proc Natl Acad Sci USA 1977;74:3090-4. Back to cited text no. 31 |
| 32. | Stanley EF, Drachman DB. Effect of myasthenic immunoglobulin on acetylcholine receptors of intact mammalian neuromuscular junctions. Science 1978;200:1285-7. Back to cited text no. 32 [PUBMED] [FULLTEXT] |
| 33. | Wilson S, Vincent A, Newsom-Davis J. Acetylcholine receptor turnover in mice with passively transferred myasthenia gravis: I, Receptor degradation. J Neurol Neurosurg Psychiatry 1983;46:377-82. Back to cited text no. 33 [PUBMED] [FULLTEXT] |
| 34. | Wilson S, Vincent A, Newsom-Davis J. Acetylcholine receptor turnover in mice with passively transferred myasthenia gravis: II, Receptor synthesis. J Neurol Neurosurg Psychiatry 1983;46:383-7. Back to cited text no. 34 [PUBMED] [FULLTEXT] |
| 35. | Guyon T, Wakkach A, Poea S, Mouly V, Klingel-Schmitt I, Levasseur P, et al. Regulation of acetylcholine receptor gene expression in human myasthenia gravis muscles: Evidences for a compensatory mechanism triggered by receptor loss. J Clin Invest 1998;102:249-63. Back to cited text no. 35 [PUBMED] [FULLTEXT] |
| 36. | Engel AG. Myasthenia gravis and myasthenic syndromes. Ann Neurol 1984;16:519-34. Back to cited text no. 36 [PUBMED] |
| 37. | Sahashi K, Engel AG, Lambert EH, Howard FM Jr. Ultrastructural localization of the terminal and lytic ninth complement component (C9) at the motor end-plate in myasthenia gravis. J Neuropathol Exp Neurol 1980;39:160-72. Back to cited text no. 37 [PUBMED] |
| 38. | Engel AG, Arahata K. The membrane attack complex of complement at the endplate in myasthenia gravis. Ann N Y Acad Sci 1987;505:326-32. Back to cited text no. 38 [PUBMED] [FULLTEXT] |
| 39. | Ruff RL, Lennon VA. Endplate voltage-gated sodium channels are lost in clinical and experimental myasthenia gravis. Ann Neurol 1998;43:370-9. Back to cited text no. 39 [PUBMED] |
| 40. | Kaminski HJ, Li Z, Richmonds C, Ruff R.L, Kusner L. Susceptibility of ocular tissues to autoimmune diseases. Ann N Y Acad Sci 2003;998:362-74. Back to cited text no. 40 |
| 41. | Plomp JJ, Van Kempen GT, De Baets MB, Graus YM, Kuks JB, Molenaar PC. Acetylcholine release in myasthenia gravis: Regulation at single end-plate level. Ann Neurol 1995;37:627-36. Back to cited text no. 41 [PUBMED] |
| 42. | Schluep M, Willcox N, Vincent A, Dhoot GK, Newsom-Davis J. Acetylcholine receptors in human thymic myoid cells in situ: An immunohistological study. Ann Neurol 1987;22:212-22. Back to cited text no. 42 [PUBMED] |
| 43. | Mesnard-Rouiller L, Bismuth J, Wakkach A, Poea-Guyon S, Berrih-Aknin S. Thymic myoid cells express high levels of muscle genes. J Neuroimmunol 2004;148:97-105. Back to cited text no. 43 |
| 44. | Scadding GK, Vincent A, Newsom-Davis J, Henry K. Acetylcholine receptor antibody synthesis by thymic lymphocytes: Correlation with thymic histology. Neurology 1981;31:935-43. Back to cited text no. 44 [PUBMED] |
| 45. | Vincent A, Newsom-Davis J, Newton P, Beck N. Acetylcholine receptor antibody and clinical response to thymectomy in myasthenia gravis. Neurology 1983;33:1276-82. Back to cited text no. 45 [PUBMED] |
| 46. | Leite MI, Jones M, Strφbel P, Marx A, Gold R, Niks E, et al. Myasthenia gravis thymus: Complement vulnerability of epithelial and myoid cells, complement attack on them, and correlations with autoantibody status. Am J Pathol 2007;171:893-905. Back to cited text no. 46 |
| 47. | Giraud M, Taubert R, Vandiedonck C, Ke X, Levi-Strauss M, Pagani F, et al. An IRF8-binding promoter variant and AIRE control CHRNA1 promiscuous expression in thymus. Nature 2007;448:934-7. Back to cited text no. 47 |
| 48. | Buckley C, Douek D, Newsom-Davis J, Vincent A, Willcox N. Mature, long-lived CD4+ and CD8+ T cells are generated by the thymoma in myasthenia gravis. Ann Neurol 2001;50:64-72. Back to cited text no. 48 [PUBMED] |
| 49. | Strobel P, Rosenwald A, Beyersdorf N, Kerkau T, Elert O, Murumagi A, et al. Selective loss of regulatory T cells in thymomas. Ann Neurol 2004;56:901-4. Back to cited text no. 49 |
| 50. | Mossman S, Vincent A, Newsom-Davis J. Myasthenia gravis without acetylcholine-receptor antibody: A distinct disease entity. Lancet 1986;1:116-9. Back to cited text no. 50 [PUBMED] [FULLTEXT] |
| 51. | Burges J, Vincent A, Molenaar PC, Newsom-Davis J, Peers C, Wray D. Passive transfer of seronegative myasthenia gravis to mice. Muscle Nerve 1994;17:1393-400. Back to cited text no. 51 [PUBMED] |
| 52. | Kong XC, Barzaghi P, Ruegg MA. Inhibition of synapse assembly in mammalian muscle in vivo by RNA interference. EMBO Rep 2004;5:183-8. Back to cited text no. 52 [PUBMED] [FULLTEXT] |
| 53. | Evoli A, Tonali PA, Padua L, Monaco ML, Scuderi F, Batocchi AP, et al. Clinical correlates with anti-MuSK antibodies in generalized seronegative myasthenia gravis. Brain 2003;126:2304-11. Back to cited text no. 53 [PUBMED] [FULLTEXT] |
| 54. | Farrugia ME, Robson MD, Clover L, Anslow P, Newsom-Davis J, Kennett R, et al. MRI and clinical studies of facial and bulbar muscle involvement in MuSK antibody-associated myasthenia gravis. Brain 2006;129:1481-92. Back to cited text no. 54 [PUBMED] [FULLTEXT] |
| 55. | Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med 2001;7:365-8. Back to cited text no. 55 [PUBMED] [FULLTEXT] |
| 56. | Vincent A, Leite MI. Neuromuscular junction autoimmune disease: Muscle specific kinase antibodies and treatments for myasthenia gravis. Curr Opin Neurol 2005;18:519-25. Back to cited text no. 56 [PUBMED] [FULLTEXT] |
| 57. | Leite MI, Strobel P, Jones M, Micklem K, Moritz R, Gold R, et al. Fewer thymic changes in MuSK antibody-positive than in MuSK antibody-negative MG. Ann Neurol 2005;57:444-8. Back to cited text no. 57 |
| 58. | Niks EH, Kuks JB, Roep BO, Haasnoot GW, Verduijn W, Ballieux BE, et al. Strong association of MuSK antibody-positive myasthenia gravis and HLA-DR14-DQ5. Neurology 2006;66:1772-4. Back to cited text no. 58 [PUBMED] [FULLTEXT] |
| 59. | McConville J, Farrugia ME, Beeson D, Kishore U, Metcalfe R, Newsom-Davis J, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol 2004;55:580-4. Back to cited text no. 59 [PUBMED] [FULLTEXT] |
| 60. | Shiraishi H, Motomura M, Yoshimura T, Fukudome T, Fukuda T, Nakao Y, et al. Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol 2005;57:289-93. Back to cited text no. 60 [PUBMED] [FULLTEXT] |
| 61. | Nemoto Y, Kuwabara S, Misawa S, Kawaguchi N, Hattori T, Takamori M, et al. Patterns and severity of neuromuscular transmission failure in seronegative myasthenia gravis. J Neurol Neurosurg Psychiatry 2005;76:714-8. Back to cited text no. 61 [PUBMED] [FULLTEXT] |
| 62. | Farrugia ME, Kennett RP, Newsom-Davis J, Hilton-Jones D, Vincent A. Single-fiber electromyography in limb and facial muscles in muscle-specific kinase antibody and acetylcholine receptor antibody myasthenia gravis. Muscle Nerve 2006;33:568-70. Back to cited text no. 62 [PUBMED] [FULLTEXT] |
| 63. | Spreadbury I, Kishore U, Beeson D, Vincent A. Inhibition of acetylcholine receptor function by seronegative myasthenia gravis non-IgG factor correlates with desensitisation. J Neuroimmunol 2005;162:149-56. Back to cited text no. 63 [PUBMED] [FULLTEXT] |
| 64. | O'Neill JH, Murray NM, Newsom-Davis J. The Lambert-Eaton myasthenic syndrome: A review of 50 cases. Brain 1998;111:577-96. Back to cited text no. 64 |
| 65. | Wirtz PW, Willcox N, Roep BO, Lang B, Wintzen AR, Newsom-Davis J, et al. HLA-B8 in patients with the Lambert-Eaton myasthenic syndrome reduces likelihood of associated small cell lung carcinoma. Ann N Y Acad Sci 2003;998:200-1. Back to cited text no. 65 [PUBMED] [FULLTEXT] |
| 66. | Lang B, Newsom-Davis J, Wray D, Vincent A, Murray N. Autoimmune aetiology for myasthenic (Eaton-Lambert) syndrome. Lancet 1981;2:224-6. Back to cited text no. 66 [PUBMED] |
| 67. | Lambert EH, Elmqvist D. Quantal components of end-plate potentials in the myasthenic syndrome. Ann N Y Acad Sci 1971;183:183-99. Back to cited text no. 67 [PUBMED] [FULLTEXT] |
| 68. | Engel AG. Review of evidence for loss of motor nerve terminal calcium channels in Lambert-Eaton myasthenic syndrome. Ann N Y Acad Sci 1991;635:246-58. Back to cited text no. 68 [PUBMED] [FULLTEXT] |
| 69. | Fukunaga H, Engel AG, Lang B, Newsom-Davis J, Vincent A. Passive transfer of Lambert-Eaton myasthenic syndrome with IgG from man to mouse depletes the presynaptic membrane active zones. Proc Natl Acad Sci U S A 1983;80:7636-40. Back to cited text no. 69 [PUBMED] [FULLTEXT] |
| 70. | Fukuoka T, Engel AG, Lang B, Newsom-Davis J, Vincent A. Lambert-Eaton myasthenic syndrome: II, Immunoelectron microscopy localization of IgG at the mouse motor end-plate. Ann Neurol 1987;22:200-11. Back to cited text no. 70 [PUBMED] |
| 71. | Peers C, Johnston I, Lang B, Wray D. Cross-linking of presynaptic calcium channels: A mechanism of action for Lambert-Eaton myasthenic syndrome antibodies at the mouse neuromuscular junction. Neurosci Lett 1993;153:45-8. Back to cited text no. 71 [PUBMED] |
| 72. | Uchitel OD, Protti DA, Sanchez V, Cherksey BD, Sugimori M, Llinas R. P-type voltage-dependent calcium channel mediates presynaptic calcium influx and transmitter release in mammalian synapses. Proc Natl Acad Sci U S A 1992;89:3330-3. Back to cited text no. 72 |
| 73. | Motomura M, Johnston I, Lang B, Vincent A, Newsom-Davis J. An improved diagnostic assay for Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry 1995;58:85-7. Back to cited text no. 73 [PUBMED] [FULLTEXT] |
| 74. | Lennon VA, Kryzer TJ, Griesmann GE, O'Suilleabhain PE, Windebank AJ, Woppmann, et al. Calcium-channel antibodies in the Lambert-Eaton syndrome and other paraneoplastic syndromes. N Engl J Med 1995;332:1467-74. Back to cited text no. 74 |
| 75. | Graus F, Lang B, Pozo-Rosich P, Saiz A, Casamitjana R, Vincent A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology 2002;59:764-6. Back to cited text no. 75 [PUBMED] [FULLTEXT] |
| 76. | Pinto A, Gillard S, Moss F, Whyte K, Brust P, Williams M, et al. Human autoantibodies specific for the alpha1A calcium channel subunit reduce both P-type and Q-type calcium currents in cerebellar neurons. Proc Natl Acad Sci U S A 1998;95:8328-33. Back to cited text no. 76 [PUBMED] [FULLTEXT] |
| 77. | Giovannini F, Sher E, Webster R, Boot J, Lang B. Calcium channel subtypes contributing to acetylcholine release from normal, 4-aminopyridine-treated and myasthenic syndrome auto-antibodies-affected neuromuscular junctions. Br J Pharmacol 2002;136:1135-45. Back to cited text no. 77 [PUBMED] [FULLTEXT] |
| 78. | Waterman SA, Lang B, Newsom-Davis J. Effect of Lambert-Eaton myasthenic syndrome antibodies on autonomic neurons in the mouse. Ann Neurol 1997;42:147-156. Back to cited text no. 78 [PUBMED] |
| 79. | Newsom-Davis J, Mills KR. Immunological associations of acquired neuromyotonia (Isaacs' syndrome): Report of five cases and literature review. Brain 1993;116:453-69. Back to cited text no. 79 [PUBMED] [FULLTEXT] |
| 80. | Hart IK, Maddison P, Newsom-Davis J, Vincent A, Mills KR. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain 2002;125:1887-95. Back to cited text no. 80 [PUBMED] [FULLTEXT] |
| 81. | Turner MR, Madkhana A, Ebers GC, Clover L, Vincent A, McGavin G, et al. Wasp sting induced autoimmune neuromyotonia. J Neurol Neurosurg Psychiatry 2006:77:704-5. Back to cited text no. 81 |
| 82. | Sinha S, Newsom-Davis J, Mills K, Byrne N, Lang B, Vincent A. Autoimmune aetiology for acquired neuromyotonia (Isaacs' syndrome). Lancet 1991;338:75-7. Back to cited text no. 82 [PUBMED] |
| 83. | Shillito P, Molenaar PC, Vincent A, Leys K, Zheng W, van den Berg RJ, et al. Acquired neuromyotonia: Evidence for autoantibodies directed against K+ channels of peripheral nerves. Ann Neurol 1995;38:714-22. Back to cited text no. 83 [PUBMED] |
| 84. | Hart IK, Waters C, Vincent A, Newland C, Beeson D, Pongs O, et al. Autoantibodies detected to expressed K+ channels are implicated in neuromyotonia. Ann Neurol 1997;41:238-46. Back to cited text no. 84 [PUBMED] |
| 85. | Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci 2004;24:1236-44. Back to cited text no. 85 [PUBMED] [FULLTEXT] |
| 86. | Kleopa KA, Elman LB, Lang B, Vincent A, Scherer SS. Neuromyotonia and limbic encephalitis sera target mature Shaker-type K+ channels: Subunit specificity correlates with clinical manifestations. Brain 2006;129:1570-84. Back to cited text no. 86 [PUBMED] [FULLTEXT] |
| 87. | Sonoda Y, Arimura K, Kurono A, Suehara M, Kameyama M, Minato S, et al. Serum of Isaacs' syndrome suppresses potassium channels in PC-12 cell lines. Muscle Nerve 1996;19:1439-46. Back to cited text no. 87 [PUBMED] |
| 88. | Tomimitsu H, Arimura K, Nagado T, Watanabe O, Otsuka R, Kurono A, et al. Mechanism of action of voltage-gated K+ channel antibodies in acquired neuromyotonia. Ann Neurol 2004;56:440-4. Back to cited text no. 88 [PUBMED] [FULLTEXT] |
| 89. | Arimura K, Sonoda Y, Watanabe O, Nagado T, Kurono A, Tomimitsu H, et al. Isaacs' syndrome as a potassium channelopathy of the nerve. Muscle Nerve 2002;11:S55-8. Back to cited text no. 89 [PUBMED] |
| 90. | Liguori R, Vincent A, Clover L, Avoni P, Plazzi G, Cortelli P, et al. Morvan's syndrome: Peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain 2001;124:2417-26. Back to cited text no. 90 [PUBMED] [FULLTEXT] |
| 91. | Irani SR, Buckley C, Vincent A, Cockerell C, Rudge-P, Johnson MR, et al. Immunotherapy-responsive seizure-like episodes with potassium channel antibodies. Neurology 2008 in press. Back to cited text no. 91 |
Copyright 2008 - Neurology India
The following images related to this document are available:
Photo images
[ni08081f2.jpg]
[ni08081t1.jpg]
[ni08081f3.jpg]
[ni08081f1.jpg]
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}