
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 56, Num. 3, 2008, pp. 314-324
|
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 314-324
Review Article
Distal myopathies a review: Highlights on distal myopathies with rimmed vacuoles
Malicdan May Christine V, Nonaka Ikuya
Department of Neuromuscular Research, National Institutes of Neurosciences, National Center of Neurology and Psychiatry, Tokyo
Correspondence Address:4-1-1 Ogawahigashi-cho, Kodaira 187-8502 Tokyo
nonaka@ncnp.go.jp
Date of Acceptance: 19-Sep-2008
Code Number: ni08082
Abstract Distal myopathies are a group of heterogeneous disorders classified into one broad category due to the presentation of weakness involving the distal skeletal muscles. The recent years have witnessed increasing efforts to identify the causative genes for distal myopathies. The identification of few causative genes made the broad classification of these diseases under "distal myopathies" disputable and added some enigma to why distal muscles are preferentially affected. Nevertheless, with the clarification of the molecular basis of specific conditions, additional clues have been uncovered to understand the mechanism of each condition. This review will give a synopsis of the common distal myopathies, presenting salient facts regarding the clinical, pathological, and molecular aspects of each disease. Distal myopathy with rimmed vacuoles, or Nonaka myopathy, will be discussed in more detail.
Keywords: Amyloid, hIBM, sialic acid
Introduction
Although proximal muscles of the extremities are predominantly affected in most primary myopathies including muscular dystrophies, there are several diseases preferentially involving distal muscles from the early stage of the disease and thus have been labeled as distal myopathies. Classification of the distal myopathies was therefore a matter of dispute; most in the past were classified on the age of onset of the disease and mode of inheritance, [1],[2],[3],[4],[5] though recent studies have shown a large list of diseases based on molecular biologic aspects. [6] Despite that the term "distal myopathy" may not be exactly accurate, as some conditions included in this classification actually are characterized by dystrophic changes in the muscle, it is maintained for historical purposes and clinical classification.
Since the discovery of the gene loci for a number of distal myopathies, several diseases previously categorized as different disorders have now proven to be the same or allelic disorders (e.g. distal myopathy with rimmed vacuoles and hereditary inclusion body myopathy, Miyoshi myopathy and limb-girdle muscular dystrophy type 2B (LGMD 2B).
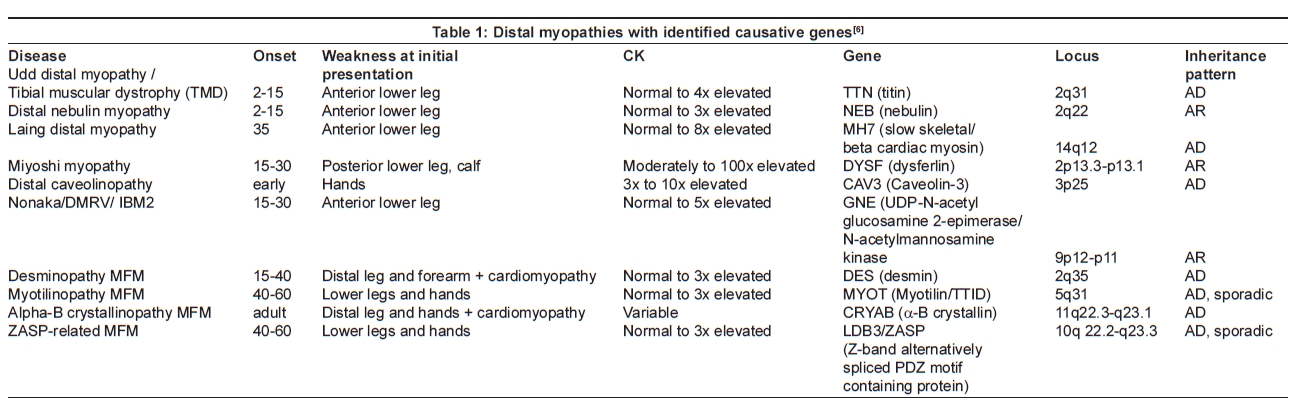
This review will focus on the most commonly known and distinct distal myopathies, using a simple classification: distal myopathies with known molecular defects and distal myopathies with unknown causative genes [Table 1]. The identification of the genes involved in distal myopathies has broadened this classification into sub-categories as to the location of encoded proteins: sarcomere (titin, myosin); plasma membrane (dysferlin, caveolin); cytoskeleton (rare; includes desmin, myotilin, αB-crystallin, ZASP, filamin C, nebulin); and cytosol (GNE) [Table 2].
Tibial Muscular Dystrophy (late onset, Type 2)
Clinical and pathologic characteristics
Udd et al. [7] first reported 66 patients from several Finnish families who had an autosomal dominantly inherited late adult onset distal leg myopathy with weakness confined mainly to the anterior tibial muscles. They named the disorder tibial muscular dystrophy (TMD), because they thought that the muscle pathology was similar to that seen in muscular dystrophy, including muscle fiber necrosis and fat tissue replacement, but serum creatine kinase (CK) levels were normal or slightly elevated.
The clinical phenotype presents after the age of 35 years, with almost selective involvement of the anterior tibial muscles and long toe extensors initially, resulting in moderate foot drop in 10-20 years. The disease severity varies from the absence of symptoms to marked difficulty in walking. Weakness may initially be asymmetric; progression of symptoms is slow, whereby patients could develop proximal leg muscles at the age of 70. Affected individuals are usually ambulant; however, elderly patients may need walking aid. Cardiac, facial, and respiratory muscles are usually not affected. [6],[7]
Biopsies of the affected tibialis anterior muscles show dystrophic changes of variable severity, including marked variation in fiber size, occasional fiber necrosis and regeneration, increased number of fibers with internal nuclei, and fiber splitting. Rimmed vacuoles (RVs) are present in the majority of patients, especially in the early stages, but muscle fibers are subsequently replaced by fat and fibrous tissue when the vacuoles are no longer discernible. Thus, the presence of RVs is not mandatory for this diagnosis. There was no immunoreactivity for tau, beta-amyloid or beta-amyloid precursor protein in the vacuolated fibers, in contrast to their positive immunoreactivity in distal myopathy with rimmed vacuoles (DMRV)/hereditary inclusion body myopathy (hIBM). These RVs in TMD are usually not membrane-bound and thus are not thought to fulfill the morphologic criteria of autophagic vacuoles, even though the vacuolar space contains numerous vesicles compatible with lysosomal autophagic components. [6]
Molecular aspects
The gene in TMD has been mapped to Chromosome 2q31 [8],[9] and mutations were found in a gene encoding a structural protein titin. [10] Deletion of 11 bp was initially found in the last exon encoding titin; later heterozygous missense mutations were also documented. Titin is the largest single polypeptide protein and each molecule spans over one half of the sarcomere from Z-disk to M-line interacting both with thin filaments and thick filaments. [6] Titin interacts repeatedly with myosin filaments in the A-band region; this gives the contractile system a strong positional fixation and keeps myosin-thick filaments always centered in the sarcomere. [6] Although TMD was initially identified among Finnish patients, recent reports show that this disease is also found in the French [11] and Belgian [12] population. After the discovery of TTN as the gene responsible for TMD, mutations in the same gene have also been shown to induce proximal muscle involvement of limb-girdle muscular dystrophy (LGMD 2J). [13]
Laing Distal Myopathy
Clinical and pathologic characteristics
Laing distal myopathy (MPD1) was originally described by Gowers in 1902, and was later reported by Laing et al. [14] It is an autosomal dominant distal myopathy with an onset as young as four or five years, although the disease onset varied from four to 25 years. It is a distinct condition characterized by weakness affecting the anterior compartment of the lower leg and selective involvement of the toe extensors, giving rise to the characteristic hanging big toe sign. The disease is slowly progressive, i.e. a patient has been reported to maintain independent ambulation 23 years after the initial investigation, whereby patients gradually develop weakness of finger flexors and proximal muscles including neck flexors, shoulder, trunk, facial and tongue muscles. Cardiomyopathy is rare and severe respiratory problems have not been described. Serum CK levels are mildly elevated.
The pathological features are variable and there are no pathognomonic diagnostic features. [14],[15] In general, fiber size distribution appears to be bimodal. Type I fibers are atrophic in 50% of the population and express both slow- and fast-type myosin. In tibialis anterior muscles, virtually all muscles abnormally express fast-type myosin. RVs are found in a minority of patients with MPD1, but not prominent. Immunohistochemical staining for slow and fast myosin showed co-expression of slow and fast myosin in some Type I fibers, possibly indicating fiber type switching. This finding seems to be a useful aid to diagnosis.
Molecular aspects
The gene for this myopathy is mapped to chromosome 14 and the mutations are found in the slow myosin heavy chain gene MYH7 , [16] hence it is sometimes know as myosinopathy. Mutations have been discovered on the MYH7 tail region, which is physically located in the M-line of the sarcomere, and where it was described to interact with myomesin and titin. Mutations in MYH7 have also been reported in patients with hyaline body myopathy, suggesting that this myopathy is allelic with myosinopathy.
Distal Nebulin Myopathy
Clinical and pathological characteristics
Recently, four families of Finnish decent were described to have an early onset distal myopathy with remarkable involvement of the ankle dorsiflexors (foot drop); other muscles severely involved include finger extensors and neck flexors. In some of the older patients, mild proximal muscle weakness was noted. Moderate facial weakness was seen in few patients. [17]
The muscle biopsy can generally be described as myopathic, although the severity of the pathology varied remarkably. Chronic atrophy was suggested by the presence of large hypertrophic fibers with increase in internal nuclei and nuclear clumps that express neonatal MHC. Nemaline bodies were not observed on light microscopy histochemistry, but were seen in toluidine blue-stained semi-thin epon sections of some patients.
Molecular aspects
Extensive analysis of patients revealed an abnormal SSCP band in the nebulin gene (NEB) , in two of the four families. Gene sequencing revealed homozygous mutations in NEB . [17] This was rather surprising because recessive mutations in NEB have been associated with nemaline myopathy, clinical picture of which is similar to that of most congenital myopathy and presents with proximal myopathy. Actually, in nemaline myopathy, most of the mutations identified were either nonsense, frameshift, or splice-site mutations, while mutations in the distal myopathy phenotype are missense. Thus Wallgren-Petterson et al. concluded that homozygozity for NEB missense mutations causes a distinct type of recessively inherited distal myopathy, [17] albeit rarely.
Autosomal Recessive distal Muscular Dystrophy (Miyoshi myopathy; early adult onset, Type 2)
Clinical and pathological characteristics
Miyoshi myopathy (MM) is an adult-onset autosomal recessive condition characterized by preferential gastrocnemius muscle involvement and dystrophic muscle pathology. [18],[19] MM seems to be widely distributed throughout the world, because many similar patients have been described from various countries. [20],[21],[22]
The symptoms and the onset of the disease can be variable, but most patients become aware of difficulty in walking in early adulthood, from 20 to 40 years. In the early stages of the disease, patients have muscle atrophy and weakness in the distal parts of legs, predominantly of the gastrocnemius and soleus muscles, and therefore have difficulty in standing on tip-toe. The disease subsequently progresses rather rapidly and muscle atrophy/weakness becomes more prominent and may spread to the proximal muscles. A few patients become non-ambulant 10-20 years after onset of the disease. Cardiac and respiratory muscles are not involved. Serum CK levels are elevated to 20-100 times the normal value. Muscle computed tomography (CT) shows preferential soleus, gastrocnemius and occasionally paraspinal muscle involvement. Despite the characteristic involvement of the posterior lower leg muscles, there is a variant of MM which peculiarly involved the tibialis anterior among a Spanish population, and hence was called distal myopathy with anterior tibialis involvement (DMAT). [23]
Muscle biopsies show dystrophic changes with active fiber necrosis and regeneration, interstitial fibrosis, and fat tissue replacement, [18],[19] similar to those seen in Duchenne or limb-girdle muscular dystrophy. Inflammatory cellular infiltration is commonly seen and could sometimes lead to misdiagnosis of polymyositis. [24]
Molecular aspects
The causative gene is DYSF gene in Chromosome 2p13. [25]
Mutations are variable and include insertions, deletions, altered splicing and point mutations. The exact function of dysferlin remains unknown, but it is thought to allow rapid membrane resealing of membranes disrupted by mechanical stress. Interestingly, patients with LGMD2B with gene locus at 2p13 also have mutations in DYSF, suggesting heterogeneous phenotypic expressions in the DYSF gene mutations. [26] This is not surprising, because MM patients occasionally show apparent proximal muscle involvement as the disease advances.
Caveolinopathy
Clinical and pathological characteristics
Caveolinopathy is known to cause LGMD1C, however, other phenotypes have been associated with this condition, including distal myopathy, rippling muscle disease, and hyperCKemia. [27] In the initial report of this caveolinopathy-associated distal myopathy, onset of weakness and atrophy was at 12 years of age, mainly involving the intrinsic muscles in the hands and feet, without involvement of the proximal muscles. The progression was slow. Muscle biopsy showed mild variation in fiber size, increased number of internalized nuclei, and predominance of Type 1 fibers.
Molecular aspects
The causative gene is CAV3 , which encodes a protein that is implicated in the development of the T-tubule system in the skeletal muscle. Mutations are mostly heterozygous missense mutations, but one deletion mutation was also identified. Like in all caveolinopathies, the expression of caveolin 3 is reduced, however, it should be noted that caveolin 3 expression can also be secondarily reduced in some muscular dystrophies like sarcoglycanopathies and dytrophinopathies, underscoring the importance of genetic screening for final diagnosis.
Myofibrillar Myopathy
The term myofibrillar myopathy (MFM) was proposed by AG Engel′s group in 1996 as a non-committal term for a pathologic pattern of myofibrillar dissolution associated with accumulation of myofibrillar degradation products and ectopic expression of multiple proteins including desmin, αB-crystallin, dystrophin and amyloid material. [28],[29] This condition had previously been labeled as spheroid body myopathy, cytoplasmic body myopathy, Mallory body myopathy and desmin storage myopathy, among other names. In this review, some MFMs are included as patients can present with distal weakness, but it is important to note that both distal and proximal muscles can be involved. [30] In patients with MFM, mutations were found in DES (desmin), CRYAB (αB-crystallin), MYOT (myotilin), LDB3 (Z-band alternatively spliced PDZ-containing protein; ZASP), and FLNC (filamin C). Accordingly, although MFM is morphologically distinct it is genetically and clinically heterogeneous. [30]
Only two of 63 patients had distal myopathy. Most patients with mutations in DES had dilated cardiomyopathy and generalized muscle weakness. [31] Although many patients with distal myopathy with desmin accumulation have been reported in the literature, [32] they were probably not common in desmin myopathy with DES mutation.
Distal dominant involvement seems to be rare in patients with CRYAB [33] and ZASP [34] mutations. Interestingly, a family with autosomal dominantly inherited distal myopathy first described by Markesbery et al. [35] is now proven to be caused by the ZASP mutation A165V. [36] Haplotype studies in this family and in five other families with European ancestry carrying the identical A165V mutation share common markers at the locus suggesting the existence of a founder mutation. Further study is necessary to determine whether ZASP gene mutation commonly causes distal myopathy.
MYOT, the gene associated with autosomal dominantly inherited LGMD1A, [37],[38] also causes distal myopathy. [39],[40],[41],[42] Myotilin (myofibrillar protein with titin-like Ig domain) is a 57 kDa Z-disc component that interacts with alpha-actinin, filamin-C, FATZ (calsarcin) and actin. The onset of distal myopathy with myotilin gene mutation is in late adulthood ranging mostly from 50-75 years of age. In the lower legs, the soleus and gastrocnemius muscles are predominantly involved from the early stage, [40],41] extending to the anterior compartment with disease progression. [42] Electromyogram shows myopathic pattern with occasional neurogenic components. Serum CK levels are normal to slightly elevated.
Distal Myopathy with Rimmed Vacuoles (DMRV; Nonaka; early adult onset, Type 1)
Among the various forms of distal myopathies, DMRV has been regarded as a distinct disorder from both the clinical and pathologic aspects. [43],[44],[45] The disease is inherited as an autosomal recessive trait and characterized clinically by preferential muscle involvement of the anterior compartment of the lower legs, i.e. tibialis anterior muscle, beginning in young adulthood, and pathologically by the presence of RVs in muscle biopsies. We thought that distal muscle involvement was the initial important disease process, but Argov et al. [46] thought quadriceps sparing was the unique finding because the bulk and strength of quadriceps muscles were relatively preserved even in the advanced stages. Since pathologic findings are similar to sporadic inclusion body myositis (sIBM), Askanas et al. suggested the term "hereditary inclusion body myopathy (hIBM)" for these patients. [47],[48] Subsequently, the term hIBM has been widely used usually when discussing the pathologic findings and DMRV when referring to patients clinically.
Clinical and pathologic findings
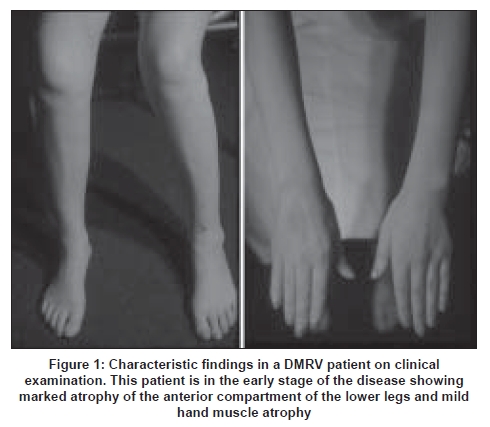
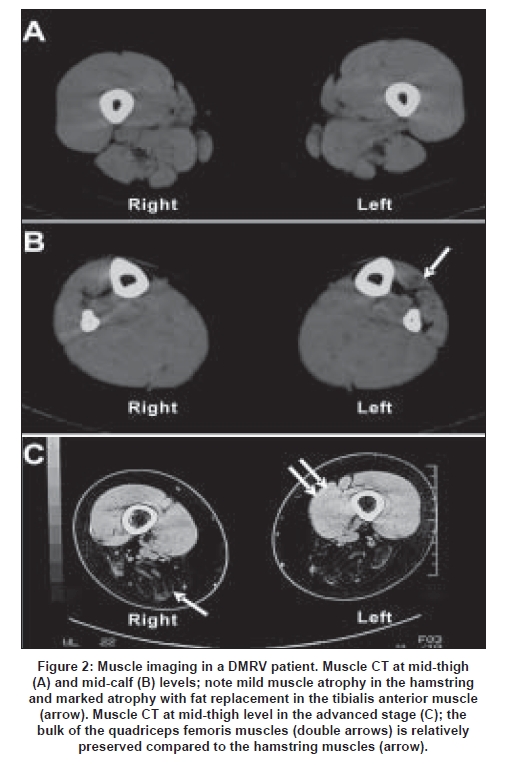
The age at onset of the disease ranges from 15 to 40 years, averaging 26 years. [43],[45] Since the tibialis anterior muscle is preferentially involved from the early stage [Figure 1], the initial symptom is gait disturbance, usually with difficulty in climbing stairs and running. The disease is rather rapidly progressive. [48],[49] Patients become non-ambulant between 26 and 57 years, average of 12 years after the onset of the disease. [43],[49] Cardiac and respiratory muscles are less involved. Although the anterior tibial muscle is most significantly affected, gastrocnemius, hamstrings, paraspinal and sternocleidomastoid muscles are also involved from an early stage when one examines muscles by CT [Figure 2A] and [Figure 2B]. Even in the advanced stages, quadriceps muscles are relatively spared [Figure 2C]. Serum CK levels are normal to mildly elevated.
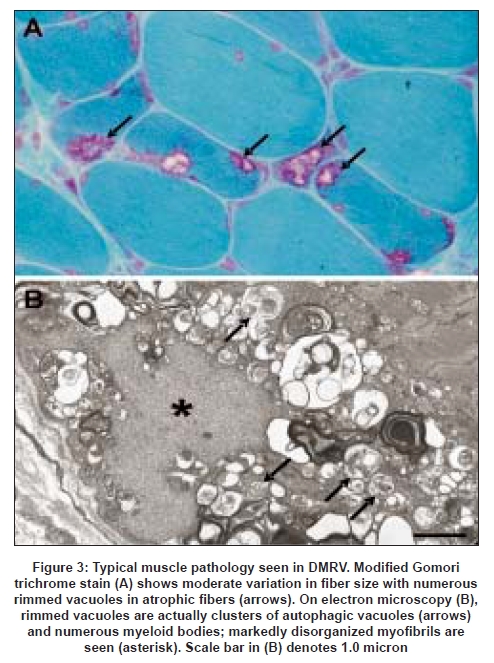
In all muscle biopsies, there are myopathic changes with variation in size in both Type 1 and 2 fibers. RVs are present predominantly in atrophic fibers which are occasionally aggregated forming small groups [Figure 3A]. [43],[45] Necrotic and regenerating fibers can be rarely seen. Type 1 fibers tend to predominate as the disease progresses. [43]
Process of muscle fiber degeneration
RV formation is thought to be the primary pathologic event which induces muscle fiber atrophy and loss in DMRV, but its exact significance remains uncertain. By electron microscopy, the RV consists of autophagic vacuoles and myeloid bodies [Figure 3B] . In the vicinity of the RVs, myofibrils are frequently disorganized, therefore degenerated contractile proteins and other cytoplasmic debris appear to activate the lysosome (autophagosome) to scavenge them. [50],[51] In the myofibrillar degeneration pathways, non-lysosomal ATP-ubiquitin proteasome proteolysis was proposed to play a role as there is increased proteasome activity in and around RVs. [52] The RV itself is not specific for DMRV, but is also found in other disorders, including chronic muscular dystrophies, metabolic disorders, myotonic dystrophy, [45],[53] oculopharyngeal muscular dystrophy, oculopharyngodistal myopathy, and Marinesco-Sjogren syndrome. [45]
RVs in muscle fibers in sIBM [48] and DMRV [54] occasionally contain congophilic deposits, which are also positively stained with β-amyloid protein precursor and tau protein antibodies. [48],[53] This notion is further supported by the finding in the DMRV model mouse: amyloid deposits preceded myofibrillar degeneration and accumulation of the autophagic vacuoles suggesting that the autophagic phenomenon is not a primary pathologic event in DMRV. [55]
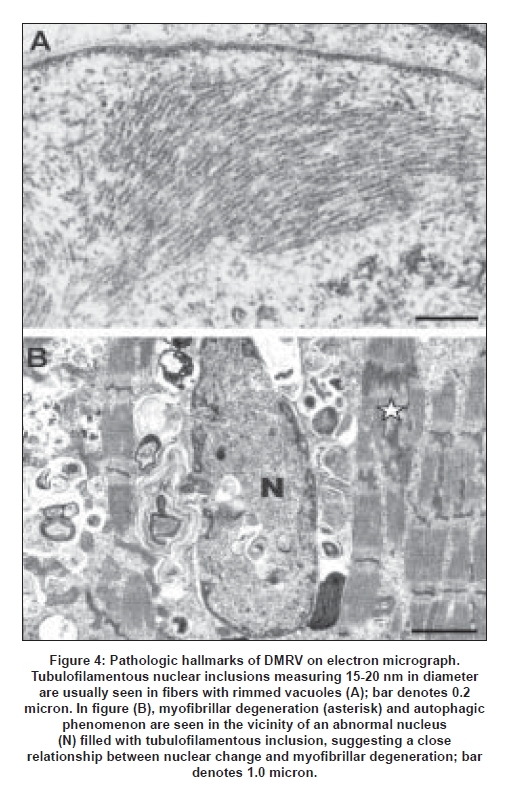
A common pathologic feature includes various nuclear inclusions and tubulofilamentous inclusions measuring 15-20 nm [Figure 4A] which are similar to 8-10 nm inclusions in oculopharyngeal muscular dystrophy. The nuclei with such inclusions are sometimes markedly degenerated and are surrounded by degenerated myofibrils, suggesting that nuclear degeneration precedes myofibrillar degeneration and autophagic phenomenon [Figure 4B]. Because some nuclei are stained positively with the TUNEL method, the nuclear change is thought to be related to apoptosis. [56]
A close relationship between nuclear change and RV formation has been suggested. In autosomal dominant inclusion body myopathy with Paget′s disease of bone and frontal dementia (IBMPFD), mutations were found in valosin-containing protein (VCP) . [57] The VCP is a polyglutamine (polyQ) interacting protein [58] and co-localizes with ubiquitin-containing inclusions in a number of neurodegenerative disorders including polyQ diseases, Parkinson, Alzheimer and Creutzfeldt-Jakob diseases. [59] Although nuclear inclusions in polyQ diseases are different from those in DMRV, nuclear degeneration probably precedes cytoplasmic vacuolar changes resulting in cell death (and probably apoptosis).
Molecular aspects
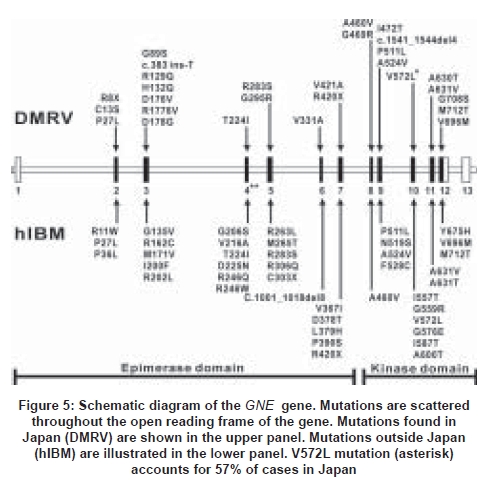
The gene for hIBM was first mapped to Chromosome 9 in 1996 [60] and in DMRV in 1997. [61] The most epoch-making event in hIBM was the discovery of mutations in UDP-N acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) , [62] a gene involved in sialic acid synthesis. GNE consists of an epimerase domain and a kinase domain [Figure 5]. The identification of GNE mutations among Japanese DMRV patients [63],[64],[65],[66] confirmed that DMRV and hIBM are the same disorders. Almost all of the mutations associated with DMRV/hIBM are missense mutations scattered throughout the open reading frame and can be found either in the epimerase domain only, in the kinase domain only, or heterozygous mutations on each domain [Figure 5]. [66]
The most common 2186T-to-C (M712T) mutation was initially thought to be restricted to Middle Eastern Jews including a large proband family analyzed by Dr Argov′s group. [62] On the other hand, the most frequent mutation in Japanese DMRV patients was the 1714C-to-G (V572L) mutation which was found in more than 50% of patients. [64],[66],[67] To date, several mutations have also been reported in other populations throughout the world. [66],[67],[68],[69]
Genotype and phenotype correlation
Although homozygous V572L mutation was thought to present with the typical clinical features of DMRV, [64] further study is necessary to clarify the genotype/phenotype correlation. A previous study reported that there was an unusual patient with proximal muscle weakness. [67] Furthermore, we had an individual with a homozygous M721T mutation with no muscle symptom, suggesting an incomplete penetrance of the disease. [66]
Two patients had inflammatory cellular infiltration in muscle biopsies mimicking the pathology of sIBM. [70,71] The onset of DMRV is in young adulthood while sIBM becomes manifest after 50 years, therefore one can differentiate the two diseases with little difficulty although there may be patients with late onset DMRV with inflammation among patients clinically diagnosed as having sIBM.
Biochemical abnormalities
The GNE enzyme is a bifunctional enzyme catalyzing two initial steps in the biosynthesis of sialic acid. Sialic acids are present at the terminal ends of glycolipid or glycoprotein on the cell surface, and are thought to contribute to glycoprotein stability, in addition to other various cellular functions. Since GNE knockout is lethal in the mouse embryo this suggests that the GNE plays a crucial role in organ synthesis. [72] Moreover, as GNE is ubiquitously expressed, mutations in the gene are thought to induce more serious disorders than DMRV in which only the muscle is affected. Nevertheless, the epimerase activity in patients was markedly reduced in white blood cells [66],[73] and lymphoblastoid cell lines, [74] suggesting that the mutations are responsible for decreased or loss of the GNE gene function. It should be noted that most mutations are missense, thus a complete loss of enzyme function may not be expected.
Altered cellular sialylation has been controversial. Hinderlich et al. found no abnormalities in patient-derived lymphoblastoid cell lines with the M712T mutation, [74] although Noguchi et al. , [73] found sialic acid levels reduced to 60-70% of control in biopsied and cultured muscle cells, and variable lectin binding reactivities from fiber to fiber. Other reports also described defective glycosylation of skeletal muscle glycoproteins, [75] including α-dystroglycan which is known to be a hyperglycosylated cell membrane protein. [76] The concept of hyposialylation was further supported by the only existing model of DMRV, the Gne knockout mouse expressing human GNE D176V mutation, wherein marked hyposialylation in serum, muscle and other organs was seen. [55]
It is still unknown how GNE mutations induce nuclear inclusion bodies and degeneration followed by amyloid accumulation and myofibrillar degeneration. O-GlcNacylation is a form not only of cytoplasmic but also of nuclear glycosylation and is present on RNA polymerase II and its associated transcriptional factors, nucleoporins etc [77] and could probably influence signal transduction. [78] Defective nuclear glycosylation may alter nuclear function which results in various defects in protein metabolism inducing amyloid deposition and protein misfolding. [55]
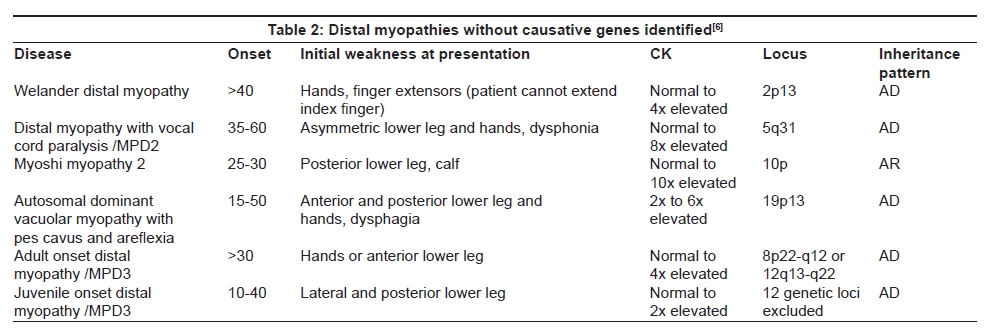
Welander Distal Myopathy (late onset, Type 1)
Welander described 249 patients from 72 pedigrees with autosomal dominant inheritance exclusively among the Swedish population. [79],[80] Onset is late, usually after the age of 40 years, and muscle weakness initially involves the distal extensor muscles of the hands and feet, and later the arms and legs. Symptoms are slowly progressive but can be detected earlier in young middle-aged relatives of the patients. [79],[81] Serum CK levels are normal or mildly elevated. On linkage analysis, Welander distal myopathy (WDM) was clearly separated from other distal myopathies and the gene was mapped to Chromosome 2p13, [82] but specific causative gene still awaits identification up to this time.
Pathological features in WDM include both myopathic and neuropathic changes, with RVs as the most striking finding. [83],[84] On electron microscopy, there is focal myofibrillar degeneration, autophagic vacuoles and nuclear inclusions with tubulofilamentous structure measuring 15-20 nm in diameter, [79] similar to those seen in DMRV/hIBM. RVs are abundant in muscle biopsies from patients with moderate to severe symptoms, but are absent in the early stages of the disease. It is therefore still uncertain whether RV formation is a primary pathologic event that induces muscle fiber atrophy and loss, or is a secondary change associated with the primary dystrophic process. [83] Decreased numbers of myelinated fibers were found in some sural nerve biopsies, suggesting that there is also peripheral nerve involvement. [84]
Vocal cord and Pharyngeal weakness with Autosomal Dominant Distal Myopathy (MPD2) A unique distal myopathy with vocal cord and pharyngeal weakness inherited as an autosomal dominant trait has been reported in a Caucasian family from Southern Tennessee. [85] The gene has been mapped Chromosome 5q31. The onset of the disease ranged from 35 to 57 years, averaging 45.7 years. Muscle weakness usually involves the feet and ankles, often in a peroneal distribution, and the hands. Vocal cord and pharyngeal weakness can be present at the onset of the distal extremity weakness. Serum CK levels are normal to mildly elevated. Muscle biopsies in six patients showed a noninflammatory myopathy with RVs, and groups of atrophic fibers consistent with denervation. No nuclear inclusions were found.
Oculopharyngodistal Myopathy
There are two categories of this disorder with different modes of inheritance, autosomal dominant [86],[87] and recessive. [88] Four families with autosomal dominant inheritance who were first described by Satoyoshi et al. , [86] had late onset external ophthalmoplegia, facial and bulbar muscle weakness, and distally dominant limb muscle weakness. Leg muscle weakness usually precedes ocular and pharyngeal symptoms. The disease is slowly progressive. It remains to be determined whether oculopharyngodistal myopathy is a variant form of oculopharyngeal muscular dystrophy, because distal weakness has also been described in some oculopharyngeal muscular dystrophy families. Recently, we examined five patients with the clinical characteristics of oculopharyngodistal myopathy for GCG expansion in poly(A)-binding protein nuclear 1 gene, the causative gene defect for oculopharyngeal muscular dystrophy. Since only one of our five patients had the significant GCG expansion, oculopharyngodistal myopathy is concluded to be a genetically heterogeneous disorder. [89]
Oculopharyngodistal myopathy with an autosomal recessive inheritance has been reported in two patients from a Japanese family who had anterior tibial muscle weakness beginning after the age of 40 and 50 years respectively. [88] Distal upper extremity weakness and drooping eyes gradually became evident. In their muscle biopsies, there were myopathic changes with RVs in atrophic fibers and cytoplasmic filaments measuring 16-18 nm in diameter.
Conclusion Precise diagnosis in distal myopathies remains as a challenge to clinicians, but some clues from the clinical history could be used for differential diagnosis [Figure 6]. The classification of distal myopathies would need some clarifications in the near future, especially that the molecular bases for such diseases are just starting to be clarified. More importantly, further analyses of the biological and molecular aspects of this disease in correlation to the clinical data, which are ongoing for some of the established entities , is expected to provide clues for understanding the mechanism behind why distal muscles are affected.
References
| 1. | Barohn RJ, Amato AA, Griggs RC. Overview of distal myopathies: From the clinical to the molecular. Neuromuscul Disord 1998;8:309-16. Back to cited text no. 1 |
| 2. | Griggs RC, Markesbery WR. Distal myopathies. In: Engel AG, Franzini-Armstrong C, editors. Myology. 2nd ed. New York: McGraw-Hill Inc.; 1994. p. 1246-52. Back to cited text no. 2 |
| 3. | Mastaglia FL, Lamont PJ, Laing NG. Distal myopathies. Curr Opin Neurol 2005;18:504-10. Back to cited text no. 3 |
| 4. | Nonaka I. Distal myopathies. Curr Opin Neurol 1999;2:493-9. Back to cited text no. 4 |
| 5. | Udd B, Griggs R. Distal myopathies. Curr Opin Neurol 2001;14:561-6. Back to cited text no. 5 |
| 6. | Udd B. Molecular biology of distal muscular dystrophies-sarcomeric proteins on top. Biochim Biophys Acta 2007;1772:145-58. Back to cited text no. 6 |
| 7. | Udd B, Partanen J, Halonen P, Falck B, Hakamies L, Heikkilδ H, et al. Tibial muscular dystrophy: Late adult-onset distal myopathy in 66 Finish patients. Arch Neurol 1993;50:604-8. Back to cited text no. 7 |
| 8. | Haravuori H, Mδkelδ-Bengs P, Udd B, Partanen J, Pulkkinen L, Somer H, et al. Assignment of the tibial muscular dystrophy locus to chromosome 2q31. Am J Hum Genet 1998;62:620-6. Back to cited text no. 8 |
| 9. | Udd B, Haravuori H, Kalimo H, Partanen J, Pulkkinen L, Paetau A, et al. Tibial muscular dystrophy: From clinical description to linkage on chromosome 2p31. Neuromuscul Disord 1998;8:327-32. Back to cited text no. 9 |
| 10. | Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, De Seze J, et al. Tibial muscular dystrophy is a titinopathy caused by mutations in TTN, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet 2002;71:492-500. Back to cited text no. 10 |
| 11. | de Seze J, Udd B, Haravuori H, Sablonniθre B, Maurage CA, Hurtevent JF, et al. The first European family with tibial muscular dystrophy outside the Finnish population. Neurology 1998;51:1746-8. Back to cited text no. 11 |
| 12. | Van den Bergh PY, Bouquiaux O, Verellen C, Marchand S, Richard I, Hackman P, et al. Tibial muscular dystrophy in a Belgian family. Ann Neurol 2003;54:248-51. Back to cited text no. 12 |
| 13. | Udd B, Vihola A, Sarparanta J, Richard I, Hackman P. Titinopathies and extension of the M-line mutation phenotype beyond distal myopathy and LGMD 2J. Neurology 2005;64:636-42. Back to cited text no. 13 |
| 14. | Laing NG, Laing BA, Meredith C, Wilton SD, Robbins P, Honeyman K, et al. Autosomal dominant distal myopathy: Linkage to chromosome 14. Am J Hum Genet 1995;56:422-7. Back to cited text no. 14 |
| 15. | Lamont PJ, Udd B, Mastaglia FL, de Visser M, Hedera P, Voit T, et al. Laing early onset distal myopathy: Slow myosin defect with variable abnormalities on muscle biopsy. J Neurol Neurosurg Psychiatry 2006;77:208-15. Back to cited text no. 15 |
| 16. | Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause Laing early-onset distal myopathy (MPD1). Am J Hum Genet 2004;75:703-8. Back to cited text no. 16 |
| 17. | Wallgren-Pettersson C, Lehtokari VL, Kalimo H, Paetau A, Nuutinen E, Hackman P, et al. Distal myopathy caused by homozygous missense mutations in the nebulin gene. Brain 2007;130:1465-76. Back to cited text no. 17 |
| 18. | Miyoshi K, Kawai H, Iwasa M, Kusaka K, Nishino H. Autosomal recessive distal muscular dystrophy as a new type of progressive muscular dystrophy: Seventeen cases in eight families including an autopsied case. Brain 1986;109:31-54. Back to cited text no. 18 |
| 19. | Nonaka I, Sunohara N, Satoyoshi E, Terasawa K, Yonemoto K. Autosomal recessive distal muscular dystrophy: A comparative study with distal myopathy with rimmed vacuole formation. Ann Neurol 1985;17:51-9. Back to cited text no. 19 |
| 20. | Bashir R, Britton S, Strachan T, Keers S, Vafiadaki E, Lako M, et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat Genet 1998;20:37-42. Back to cited text no. 20 |
| 21. | Cupler EJ, Bohlega S, Hessler R, McLean D, Stigsby B, Ahmad J. Miyoshi myopathy in Saudi Arabia: Clinical, electrophysiological, histopathological and radiological features. Neuromuscul Disord 1998;8:321-6. Back to cited text no. 21 |
| 22. | Linssen WH, de Visser M, Notermans NC, Vreyling JP, Van Doorn PA, Wokke JH, et al. Genetic heterogeneity in Miyoshi-type distal muscular dystrophy. Neuromuscul Disord 1998;8:317-20. Back to cited text no. 22 |
| 23. | Illa I, Serrano-Munuera C, Gallardo E, Lasa A, Rojas-Garcνa R, Palmer J, et al. Distal anterior compartment myopathy: A dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol 2001;49:130-4. Back to cited text no. 23 |
| 24. | Nagaraju K, Rawat R, Veszelovszky E, Thapliyal R, Kesari A, Sparks S, et al. Dysferlin deficiency enhances monocyte phagocytosis: A model for the inflammatory onset of limb-girdle muscular dystrophy 2B. Am J Pathol 2008;172:774-85. Back to cited text no. 24 |
| 25. | Bejaoui K, Hirabayashi K, Hentati F, Haines JL, Ben Hamida C, Belal S, et al. Linkage of Miyoshi myopathy (distal autosomal recessive muscular dystrophy) locus to chromosome 2p12-14. Neurology 1995;45:768-72. Back to cited text no. 25 |
| 26. | Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet 1998;20:31-6. Back to cited text no. 26 |
| 27. | Woodman SE, Sotgia F, Galbiati F, Minetti C, Lisanti MP. Caveolinopathies. Mutattions in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology 2004;62:538-43. Back to cited text no. 27 |
| 28. | Nakano S, Engel AG, Waclawik AJ, Emslie-Smith AM, Busis NA. Myofibrillar myopathy with abnormal foci of desmin positivity, I: Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol 1996;55:549-62. Back to cited text no. 28 |
| 29. | Engel AG. Myofibrillar myopathy. Ann Neurol 1999;46:681-3. Back to cited text no. 29 |
| 30. | Selcen D, Ohno K, Engel AG. Myofibrillar myopathy: Clinical, morphological and genetic studies in 63 patients. Brain 2004;127:439-51. Back to cited text no. 30 |
| 31. | Dalakas MC, Park KY, Semino-Mora C, Lee HS, Sivakumar K, Goldfarb LG. Desmin myopathy: A skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 2000;342:770-80. Back to cited text no. 31 |
| 32. | Goebel HH, Warlo IA. Progress in desmin-related myopathies. J Child Neurol 2000;15:565-72. Back to cited text no. 32 |
| 33. | Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann Neurol 2003;54:804-10. Back to cited text no. 33 |
| 34. | Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol 2005;57:269-76. Back to cited text no. 34 |
| 35. | Markesbery WR, Griggs RC, Leach RP, Lapham LW. Late onset hereditary distal myopathy. Neurology 1974;24:127-34. Back to cited text no. 35 |
| 36. | Griggs R, Vihola A, Hackman P, Talvinen K, Haravuori H, Faulkner G. Zaspopathy in a large classic late-onset distal myopathy family. Brain 2007;130:1477-84. Back to cited text no. 36 |
| 37. | Hauser MA, Horrigan SK, Salmikangas P, Torian UM, Viles KD, Dancel R, et al. Myotilin is mutated in limb girdle muscular dystrophy 1A. Hum Mol Genet 2000;9:2141-7. Back to cited text no. 37 |
| 38. | Hauser MA, Conde CB, Kowaljow V, Zeppa G, Taratuto AL, Torian UM, et al. myotilin Mutation found in second pedigree with LGMD1A. Am J Hum Genet 2002;71:1428-32. Back to cited text no. 38 |
| 39. | Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology 2004;62:1363-71. Back to cited text no. 39 |
| 40. | Olivι M, Goldfarb LG, Shatunov A, Fischer D, Ferrer I. Myotilinopathy: Refining the clinical and myopathological phenotype. Brain 2005;128:2315-26. Back to cited text no. 40 |
| 41. | Berciano J, Gallardo E, Domνnguez-Perles R, Gallardo E, Garcνa A, Garcνa-Barredo R, et al. Autosomal-dominant distal myopathy with a myotilin S55F mutation: Sorting out the phenotype. J Neurol Neurosurg Psychiatry 2008;79:205-8. Back to cited text no. 41 |
| 42. | Pιnisson-Besnier I, Talvinen K, Dumez C, Vihola A, Dubas F, Fardeau M, et al. Myotilinopathy in a family with late onset myopathy. Neuromuscul Disord 2006;16:427-31. Back to cited text no. 42 |
| 43. | Nonaka I, Sunohara N, Satoyoshi E, Terasawa K, Yonemoto K. Familial distal myopathy with rimmed vacuole and lamellar (myeloid) body formation. J Neurol Sci 1981;51:141-55. Back to cited text no. 43 |
| 44. | Kumamoto T, Fukuhara N, Nagashima M, Kanda T, Wakabayashi M. Distal myopathy: Histochemical and ultrastructural study. Arch Neurol 1982;39:367-71. Back to cited text no. 44 |
| 45. | Nonaka I, Murakami N, Suzuki Y, Kawai M. Distal myopathy with rimmed vacuoles. Neuromuscul Disord 1998;8:333-7. Back to cited text no. 45 |
| 46. | Argov Z, Yarom R. "Rimmed vacuole myopathy" sparing the quadriceps: A unique disorder in Iranian Jews. J Neurol Sci 1984;64:33-43. Back to cited text no. 46 |
| 47. | Askanas V, Engel WK. New advances in the understanding of sporadic inclusion-body myositis and hereditary inclusion-body myopathies. Curr Opin Rheumatol 1995;7:486-96. Back to cited text no. 47 |
| 48. | Askanas V, Engel WK. Sporadic inclusion-body myositis and hereditary inclusion-body myopathies: Current concepts of diagnosis and pathogenesis. Curr Opin Rheumatol 1998;10:543-7. Back to cited text no. 48 |
| 49. | Sunohara N, Nonaka I, Kamei N, Satoyoshi E. Distal myopathy with rimmed vacuole formation: A follow-up study. Brain 1989;112:65-83. Back to cited text no. 49 |
| 50. | Ii K, Hizawa K, Nonaka I, Sugita H, Kominami E, Katunuma N. Abnormal increases of lysosomal cysteine proteinases in rimmed vacuoles in the skeletal muscle. Am J Pathol 1986;122:193-8. Back to cited text no. 50 |
| 51. | Nishino I. Autophagic vacuolar myopathies. Curr Neurol Neurosci Rep 2003;3:64-9. Back to cited text no. 51 |
| 52. | Kumamoto T, Fujimoto S, Nagao S, Masuda T, Sugihara R, Ueyama H, et al. Proteasomes in distal myopathy with rimmed vacuoles. Int Med 1998;37:746-52. Back to cited text no. 52 |
| 53. | Murakami N, Ihara Y, Nonaka I. Muscle fiber degeneration in distal myopathy with rimmed vacuole formation. Acta Neuropathol 1995;89:29-34. Back to cited text no. 53 |
| 54. | Fukuhara N, Kumamoto T, Tsubaki T. Rimmed vacuoles. Acta Neuropathol 1980;51:229-35. Back to cited text no. 54 |
| 55. | Malicdan MC, Noguchi S, Nonaka I, Hayashi YK, Nishino I. A Gne knockout mouse expressing human GNE D176V mutation develops features similar to distal myopathy with rimmed vacuoles or hereditary inclusion body myopathy. Hum Mol Genet 2007;16:2669-82. Back to cited text no. 55 |
| 56. | Yan C, Ikezoe K, Nonaka I. Apoptotic muscle fiber degeneration in distal myopathy with rimmed vacuoles. Acta Neuropathol 2001;101:9-16. Back to cited text no. 56 |
| 57. | Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet 2004;36:377-81. Back to cited text no. 57 |
| 58. | Hirabayashi M, Inoue K, Tanaka K, Nakadate K, Ohsawa Y, Kamei Y, et al. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ 2001;8:977-84. Back to cited text no. 58 |
| 59. | Mizuno Y, Hori S, Kakizuka A, Okamoto K. Vacuole-creating protein in neurodegenerative diseases in humans. Neurosci Lett 2003;34:77-80. Back to cited text no. 59 |
| 60. | Mitrani-Rosenbaum S, Argov Z, Blumenfeld A, Seidman CE, Seidman JG. Hereditary inclusion body myopathy maps to chromosome 9p1-q1. Hum Mol Genet 1996;5:159-163. Back to cited text no. 60 |
| 61. | Ikeuchi T, Asaka T, Saito M, Tanaka H, Higuchi S, Tanaka K, et al. Gene locus for autosomal recessive distal myopathy with rimmed vacuoles maps to chromosome 9. Ann Neurol 1997;41:432-7. Back to cited text no. 61 |
| 62. | Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, et al. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet 2001;29:83-7. Back to cited text no. 62 |
| 63. | Kayashima T, Matsuo H, Satoh A, Ohta T, Yoshiura K, Matsumoto N, et al. Nonaka myopathy is caused by mutations in the UDP-N-acetylglucosamine -2-epimerase/N-acetylmannosamine kinase gene. J Hum Genet 2002;47:77-9. Back to cited text no. 63 |
| 64. | Tomimitsu H, Ishikawa K, Shimizu J, Ohkoshi N, Kanazawa I, Mizusawa H. Distal myopathy with rimmed vacuoles: novel mutations in the GNE gene. Neurology 2002;14:451-4. Back to cited text no. 64 |
| 65. | Arai A, Tanaka K, Ikeuchi T, Igarashi S, Kobayashi H, Asaka T, et al. A novel mutation in the GNE gene and a linkage disequilibrium in Japanese pedigrees. Ann Neurol 2002;52:516-9. Back to cited text no. 65 |
| 66. | Nishino I, Noguchi S, Murayama K, Driss A, Sugie K, Oya Y, et al. Distal myopathy with rimmed vacuoles is allelic to hereditary inclusion body myopathy. Neurology 2002;59:1689-93. Back to cited text no. 66 |
| 67. | Tomimitsu H, Shimizu J, Ishikawa K, Ohkoshi N, Kanazawa I, Mizusawa H. Distal myopathy with rimmed vacuoles (DMRV): New GNE mutations and splice variant. Neurology 2004;62:1607-10. Back to cited text no. 67 |
| 68. | Eisenberg I, Grabov-Nardini G, Hochner H, Korner M, Sadeh M, Bertorini T, et al. Mutations spectrum of GNE in hereditary inclusion body myopathy sparing the quadriceps. Hum Mutat 2003;21:99. Back to cited text no. 68 |
| 69. | Argov Z, Eisenberg I, Grabov-Nardini G, Sadeh M, Wirguin I, Soffer D, et al. Hereditary inclusion body myopathy: The Middle Eastern genetic cluster. Neurology 2003;60:1519-23. Back to cited text no. 69 |
| 70. | Krause S, Schlotter-Weigel B, Walter MC, Najmabadi H, Wiendl H, Müller-Hφcker J, et al. A novel homozygous missense mutation in the GNE gene of a patient with quadriceps-sparing hereditary inclusion body myopathy associated with muscle inflammation. Neuromuscul Disord 2003;13:830-4. Back to cited text no. 70 |
| 71. | Yabe I, Higashi T, Kikuchi S, Sasaki H, Fukazawa T, Yoshida K, et al. GNE mutations causing distal myopathy with rimmed vacuoles with inflammation. Neurology 2003;61:384-6. Back to cited text no. 71 |
| 72. | Schwarzkopf M, Knobeloch KP, Rohde E, Hinderlich S, Wiechens N, Lucka L, et al. Sialylation is essential for early development of in mice. Proc Natl Acad Sci USA 2002;99:5267-70. Back to cited text no. 72 |
| 73. | Noguchi S, Keira Y, Murayama K, Ogawa M, Fujita M, Kawahara G, et al. Reduction of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase activity and sialylation in distal myopathy with rimmed vacuoles. J Biol Chem 2004;279:11402-7. Back to cited text no. 73 |
| 74. | Hinderlich S, Salama I, Eisenberg I, Potikha T, Mantey LR, Yarema KJ, et al. The homozygous M712T mutation of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase results in reduced enzyme activities but not in altered overall cellular sialylation in hereditary inclusion body myopathy. FEBS Lett 2004;566:105-9. Back to cited text no. 74 |
| 75. | Saito F, Tomimitsu H, Arai K, Nakai S, Kanda T, Shimizu T, et al. A Japanese patient with distal myopathy with rimmed vacuoles: Missense mutations in the epimerase domain of the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene accompanied by hyposialylation of skeletal muscle glycoproteins. Neuromuscul Disord 2004;14:158-61. Back to cited text no. 75 |
| 76. | Huizing M, Rakocevic G, Sparks SE, Mamali I, Shatunov A, Goldfarb L, et al. Hypoglycosylation of a-dystroglycan in patients with hereditary IBM due to GNE mutations. Mol Genet Metab 2004;81:196-202. Back to cited text no. 76 |
| 77. | Snow DM, Hart GW. Nuclear and cytoplasmic glycosylation. Int Rev Cytol 1998;181:42-74. Back to cited text no. 77 |
| 78. | Wells L, Vosseller K, Hart GW. Glycosylation of nucleoplasmic proteins: Signal transduction and O-GlcNAc. Science 2001;291:2376-8. Back to cited text no. 78 |
| 79. | Borg K, Ahlberg G, Anvret M, Edstrφm L. Welander distal myopathy: An overview. Neuromuscul Disord 1998;8:115-8. Back to cited text no. 79 |
| 80. | Welander L. Myopathia distalis tarda hereditaria. Acta Med Scand 1951;141:1-124. Back to cited text no. 80 |
| 81. | Borg K, Ahlberg G, Borg J, Edstrφm L. Welander's distal myopathy: Clinical, neurophysiological and muscle biopsy observations in young and middle aged adults with early symptoms. J Neurol Neurosurg Psychiatry 1991;54:494-8. Back to cited text no. 81 |
| 82. | Ahlberg G, von Tell D, Borg K, Edstrφm L, Anvret M. Genetic linkage of Welander distal myopathy to chromosome 2p13. Ann Neurol 1999;46:399-404. Back to cited text no. 82 |
| 83. | Thornell LE, Edstrφm L, Billeter R, Butler-Browne GS, Kjφrell U, Whalen RG. Muscle fibre type composition in distal myopathy (Welander): An analysis with enzyme and immunohistochemical, gel-electrophoretic and ultrastructural techniques. J Neurol Sci 1984;65:269-92. Back to cited text no. 83 |
| 84. | Borg K, Solders G, Borg J, Edstrφm L, Kristensson K. Neurogenic involvement in distal myopathy (Welander): Histochemical and morphological observations on muscle and nerve biopsies. J Neurol Sci 1989;91:53-70. Back to cited text no. 84 |
| 85. | Feit H, Silbergleit A, Schneider LB, Gutierrez JA, Fitoussi RP, Rιyθs C, et al. Seboun Vocal cord and pharyngeal weakness with autosomal dominant distal myopathy: Clinical description and gene localization to 5q31. Am J Hum Genet 1998;63:1732-42. Back to cited text no. 85 |
| 86. | Satoyoshi E, Kinoshita M. Oculopharyngodistal myopathy. Arch Neurol 1977;34:89-92. Back to cited text no. 86 |
| 87. | Satoyoshi E, Sunohara N, Nonaka I. Distal myopathy with rimmed vacuoles, inclusion body myositis and related disorders in Japan. In: Askanas V, Serratrice G, Engel WK, editors. Inclusion body myositis and myopathies Cambridge: Cambridge Univ Press; 1998. p. 244-51. Back to cited text no. 87 |
| 88. | Uyama E, Uchino M, Chateau D, Tomι FM. Autosomal recessive oculopharyngodistal myopathy in light of distal myopathy with rimmed vacuoles and oculopharyngeal muscular dystrophy. Neuromuscul Disord 1998;8:119-25. Back to cited text no. 88 |
| 89. | Minami N, Ikezoe K, Kuroda H, Nakabayashi H, Satoyoshi E, Nonaka I. Oculopharyngodistal myopathy is genetically heterogeneous and most cases are distinct from oculopharyngeal muscular dystrophy. Neuromuscul Disord 2001;11:699-702. Back to cited text no. 89 |
Copyright 2008 - Neurology India
The following images related to this document are available:
Photo images
[ni08082t2.jpg]
[ni08082f6.jpg]
[ni08082f1.jpg]
[ni08082f4.jpg]
[ni08082f2.jpg]
[ni08082f5.jpg]
[ni08082t1.jpg]
[ni08082f3.jpg]
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}