 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
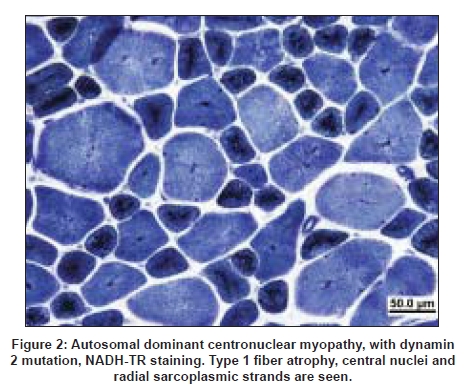
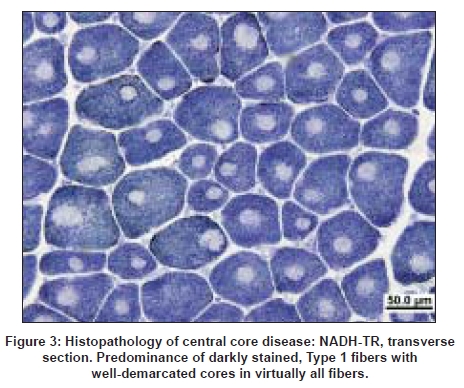
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 325-332 Review Article Myotubular/centronuclear myopathy and central core disease Fujimura-Kiyono Chieko, Racz Gabor Z, Nishino Ichizo Department of Neuromuscular Research, National Institute of Neuroscience, National Center of Neurology and Psychiatry, 4-1-1 Ogawahigashi-Cho Kodaira, Tokyo 187-8502 Date of Acceptance: 30-Aug-2008 Code Number: ni08083 Abstract The term congenital myopathy is applied to muscle disorders presenting with generalized muscle weakness and hypotonia from early infancy with delayed developmental milestones. The congenital myopathies have been classified into various categories based on morphological findings on muscle biopsy. Although the clinical symptoms may seem homogenous, the genetic basis is remarkably variable. This review will focus on myotubular myopathy, centronuclear myopathy, central core disease, and congenital neuromuscular disease with uniform Type 1 fiber, myopathies that are subjects of our ongoing examinations.Keywords: Central core disease, centronuclear myopathy, congenital myopathy, congenital neuromuscular disease with uniform Type 1 fibers, myotubular myopathy Introduction Congenital myopathies collectively denote a term used to describe a heterogeneous group of inherited muscle diseases that are typically present at birth or in the first year of life with reduced fetal movements or delay of motor milestones, as well as weakness and hypotonia. The diagnosis of a specific congenital myopathy relies predominantly on muscle pathology that reveals distinctive changes within the myofiber under light and/or electron microscopy, and not in the sarcolemmal membrane, which is the origin of the muscular dystrophies. Epidemiological data are available for congenital myopathies as a general group but not for specific diseases or conditions. The incidence of all congenital myopathies is estimated at around 6.0/100.000 live births, or one-tenth of all cases of neuromuscular disorders. [1] In our series from 1979-2007, we had 597 cases as congenital myopathy among 10332 muscle biopsies diagnosed based on muscle pathology findings. Despite their genetic heterogeneity, the clinical features have some characteristics in common. Main symptoms are generalized muscle weakness and hypotonia, typically presenting at birth or in infancy. The clinical severity is highly variable from a mild, almost asymptomatic form, to a severe form characterized by marked hypotonia at birth. Muscle weakness can be very subtle in childhood and thus can only present during adult years. Dysmorphic facies is very common due to facial muscle involvement, and when this does occur, the onset is thought to be in utero . High arched palate is often documented in almost all patients with this disease. Scoliosis and contracture of joints are seen early in childhood, especially in infantile or child onset form. On the other hand, cardiac involvement is rare and intelligence is usually normal. Clinically, it is sometimes difficult to distinguish a congenital myopathy from spinal muscular atrophy or congenital myotonic dystrophy solely based upon clinical features. Serum creatine kinase levels are within the normal range or are mildly elevated at most. Pathologically, Type 1 fiber predominance and Type 1 fiber hypotrophy are commonly seen features. In addition, the presence of characteristic structural abnormalities such as nemaline bodies, central cores and central nuclei, are the diagnostic hallmark of subtypes of congenital myopathies. Their clinical course is usually non-progressive or slowly progressive. Prognosis is often determined by respiratory, orthopedic, and bulbar involvement. Over the past decade, a number of causative genes have been identified for congenital myopathies. But up to this time, only a few of these genes have been well characterized and have clarified the pathomechanism underlying the disease. Centronuclear Myopathy Centronuclear myopathy (CNM) comprises a group of congenital myopathies which are defined pathologically by the presence of the characteristic centrally located nuclei in a large number of muscle fibers. Although the pathological findings are uniform, the clinical features and genetic background are heterogeneous. CNMs can be classified into three forms: X-linked (OMIM 310400), autosomal recessive (OMIM 255200) and autosomal dominant (OMIM 160150) forms. [2],[3] CNM was first reported under the name of "myotubular myopathy" by Spiro et al. , in 1966. [4] The term "myotubular myopathy" was used due to morphological similarities to fetal myotubes. Currently, however, the use of this term is limited to indicate only the severe infantile form, which is usually X-linked. Unlike in other congenital myopathies, ptosis and extraocular muscle involvement are frequently seen in CNMs. In addition, facial muscle involvement can be prominent, resulting in the characteristic "elongated" facies. Central or peripheral nervous systems are occasionally affected. [5] X-linked Myotubular Myopathy X-linked myotubular myopathy (XLMTM) is caused by mutations in MTM1 that encodes myotubularin and is transmitted in X-linked recessive mode. The estimated incidence is approximately one out of 50,000 males. [6] Myotubularin is a protein-tyrosine phosphatase required for muscle cell differentiation, and has been demonstrated to be a phosphatidylinositol 3-phosphate phosphatase. It is highly conserved during evolution and plays important roles in regulating intracellular membrane trafficking and vesicular transport processes. [7],[8] Myonuclear position is known to change with developmental stage. In fetal muscle, the nuclei are positioned at the center of muscle fibers, however, the majority of the nuclei are found at the periphery at birth. These pathological features resemble those of fetal muscles or myotubes, therefore XLMTM had been attributed to an arrest of myogenesis. [5] However, muscle development and differentiation were normal in MTM1 knockout mice and a myopathy with centralized nuclei was seen only after four weeks of age, clearly indicating that centrally-placed nuclei are not due to the arrest of myogenesis and that myotubularin plays a role in skeletal muscle maintenance rather than myogenesis. [9] To date, more than 200 different mutations have been identified. Originally, MTM1 mutations were associated with the severe infantile form but now we know that MTM1 mutations can also cause milder phenotypes. In the study of genotype-phenotype correlation, only 17 and seven individuals of the 116 cases were classified as mild and intermediate phenotype. [10] Furthermore, missense mutation in MTM1 was identified in a 67-year-old man, who was initially suspected to have autosomal centronuclear myopathy, and whose two grandsons also show mild phenotype. [11] Truncating mutations usually cause the severe neonatal form while non-truncating and missense mutations can lead to either mild or severe phenotype. [10],[11] Clinical characteristics A wide variety of medical complications, such as vitamin K-dependent coagulopathy, gastrointestinal bleeding, pyloric stenosis, cholestatis/cholelithiasis, scoliosis and nephrocalcinosis/kidney stones, are seen in XLMTM and can sometimes be life-threatening. Therefore, clinicians should carefully monitor patients for these potential complications. [14] Pathology Prognosis has been correlated with pathologic findings. Patients with larger myofibers tend to have better outcome and frequently become independent from respirator. In contrast, patients with small-calibered fibers continue to be ventilator-dependent and have poor prognosis. [16] The abovementioned pathological findings are very similar to those in congenital myotonic dystrophy. In fact, it is also clinically difficult to differentiate between XLMTM and congenital myotonic dystrophy, as both conditions present with severe hypotonia and muscle weakness at birth. Furthermore, recently, a newborn with spinal muscular atrophy (SMA) Type 0 showing similar clinicopathological findings with myotubular myopathy was reported. [17] Therefore, not only congenital myotonic dystrophy but also SMA Type 0 should be included in the differential diagnosis. Autosomal Dominant and Recessive Centronuclear Myopathy Autosomal dominant and recessive forms of CNM (AD-CNM, AR-CNM) have much more variable clinical characteristics, but have later onset and milder clinical course in most cases. [2] Mutations in DNM2 are responsible for autosomal dominant AD-CNM. [18] DNM2 gene encodes dynamin 2, a large guanosin triphophatase (GTPase) that plays roles in endocytosis, membrane trafficking, actin assembly and centrosome cohesion. [19],[20],[21],[22] Dynamin 2 has five domains: 1) N-terminal tripartite GTPase, 2) middle,3) pleckstrin homology, 4) GTPase effector, and 5) C-terminal proline rich domains. So far all reported DNM2 mutations associated with typical late-onset AD-CNM are located in the middle domain. In contrast, mutations associated with rare severe infantile cases are present in the pleckstrin homology domain. [23] DNM2 mutations also cause Charcot-Marie-Tooth disease Type 2B (CMT2B), for which mutations in the pleckstrin homology domain have been reported. [24],[25] The pleckstrin homology domain can function as regulated membrane-binding module that binds to inositol lipids and responds to upstream signals by targeting the host proteins to the correct cellular sites. Recently, two new genes have been added to the list: hJUMPY and BIN1. Two missense mutations in hJUMPY were identified initially in sporadic cases of CNM. [26] hJUMPY is a phosphoinositide phosphatase that shares the same substrate specificity as myotubularin. However, its pathomechanism and mode of inheritance are still unknown. BIN1 encodes amphiphysin 2 that plays a role in membrane remodeling and tubulation by interacting with dynamin 2. [27] Probably, both amphiphysin 2 and dynamin 2 play a role in normal positioning of myonuclei. More recently, a sporadic CNM patient with a de novo dominant missense mutation in the ryanodine receptor (RYR1) was reported. In this patient, quadriceps femoris muscle biopsied at age one year showed muscle pathology of CNM; however, core-like structure was seen in tibialis anterior muscle biopsied at age nine years. [28] Probably, more cases are necessary to conclude that RYR1 is the causative gene for CNM. Clinical characteristics Muscle weakness is seen with variable severity. Limb girdle and paraspinal muscles have been thought to be the most severely affected muscles in several CNM patients. However, recently, a study showed that the distribution of muscle weakness is actually more variable. [29] In fact, in DNM2-CNM, distal limb muscles, especially calf muscles and posterior thigh muscles, are more preferentially affected than proximal limb and axial muscles. [30] Achilles tendon contractures are also frequently observed. Ptosis is commonly seen in patients, as in XLMTM, but extraocular muscle weakness is only occasionally seen. [30] The central nervous system is occasionally affected and mild cognitive impairment can be seen. [4],[30] Neuropathic signs, such as absence of tendon reflexes on neurological examination and fibrillations or reduction of the compound muscle action potential (CMAP) on electrophysiological examination, are sometimes seen in DNM2-CNM patents. This suggests that there may be overlapping features between DNM2-CNM and DNM2-CMT2B phenotypes. Pathology Central Core Disease Central core disease (CCD; OMIM 117000) is characterized by clinical features of a congenital myopathy and a particular histological picture with areas of reduced oxidative activity in Type 1 (slow) muscle fibers, the "central cores" [Figure 3]. CCD was originally reported in a family with congenital hypotonia, non-progressive and non-wasting weakness of the proximal muscles of the limbs, more than half a century ago. [31] The term "central core" was introduced to indicate the area with characteristic absence of oxidative enzyme activity in the muscle fiber on NADH-TR staining; [32],[33] the absence of mitochondria can be confirmed by electron microscopy. [34] Genetic background Altogether, more than 80 point mutations have been identified in the RYR1 gene. Most mutations are missense but a few small deletions and cryptic splicing site mutations have also been documented. [37] Most cases of RYR1-associated diseases are autosomal dominant; however, rare cases are autosomal recessive. Mutations in RYR1 are also associated with malignant hyperthermia susceptibility (MHS), multi-minicore disease (MmD), and CNM, although rarely. [38] The association between CCD and MHS had been suspected early, as individuals with MHS may have central cores on muscle biopsy [39] and patients with CCD tend to have malignant hyperthermia episodes during general anesthesia induced by halogenated anesthetic agents and succinylcholine. However, MHS patients usually do not have any myopathy before developing MH syndrome. In our series, 50% of MHS cases with RYR1 mutations had cores on muscle pathology; however, none of them were typical, well-demarcated, "central cores", suggesting that those with typical CCD may not have MHS although further studies are necessary to draw any conclusion. In an extensive screening of all 106 exons in 27 Japanese CCD patients, mutations were identified in 25 (93%). [40] In fact, two cases without mutations had unusual cores, suggesting that virtually all CCD are due to RYR1 mutations. Before this report, RYR1 mutation screening for CCD cases had been limited to three "hotspots", with particular attention to the C-terminal-encoding exons; therefore only about 60% of CCD mutations had been assigned to RYR1, and the remaining were unidentified. In our series, most mutations were located in Exon 101-102, but novel mutations were found in Exon 47-48, as well as one in Exon 13. Recessive RYR1 mutations are much less frequent, and distributed along the entire gene, causing atypical CCD and MmD. [41] Zhou et al. , discovered that in half of their patients with recessive core myopathies epigenetic tissue-specific allelic silencing (leading to monoallelic transcription) unveils recessive RYR1 mutation in skeletal muscle, despite being heterozygous at the genomic level. [42] These data also suggest that imprinting by methylation is a likely mechanism for this phenomenon, and similar mechanisms can play a general role in human phenotypic heterogeneity as well as in irregularities of inheritance patterns. In addition, compound heterozygosity for RYR1 null and missense mutation has been recently identified as another important mechanism of monoallelic RYR1 expression. [43] How the mutations affect the channel function in CCD, is still controversial. One hypothesis is that these mutations lead to leaky channels, depletion of SR Ca 2+ stores and consequently muscle weakness [44] together with compensatory uptake of the excess Ca 2+ by mitochondria, which may lead to mitochondrial dysfunction and loss. [45] Alternatively, C-terminal RYR1 mutations lead to functional uncoupling of sarcolemma depolarization from release of Ca 2+ from the SR calcium stores with consequential muscle weakness. [46] Rare non-RYR1 mutations include b-myosin heavy chain mutation that cause CCD in familial hypertrophic cardiomyopathy. [47] Clinicopathologic features Serum creatine kinase (CK) levels are usually normal or only mildly elevated. However, in rare cases, it can be elevated to 6-14 times normal. [54] Muscle ultrasound often shows an increase in echogenicity even in mildly affected cases. A muscle magnetic resonance (MR) imaging study on RYR1 -related myopathies showed a consistent pattern characterized by selective involvement of the following thigh and lower leg muscles: vasti, sartorius, adductor magnus and soleus, gastrocnemius and peroneal group. [55] An earlier case report of two Japanese siblings confirmed this with the addition of the involvement of paravertebral muscles. [56] In vitro contracture test or calcium-induced calcium release test confirms the presence of MHS, and thus is recommended in CCD patients before general anesthesia. [52],[57] On muscle pathology, the core structure is most clearly demonstrated with oxidative enzyme stains such as NADH-TR, succinate dehydrogenase, and cytochrome c oxidase, because mitochondria and sarcoplasmic reticulum are absent in this area. The cores typically extend along the muscle fibers. Periodic acid-Schiff stains the central part more intensely than the periphery of the fiber. With immunostaining, in the cores myofibrillar proteins (αB-crystallin, desmin, filamin C; these are nonspecific, seen also in target fibers in various denervating diseases), and calcium-related proteins (SERCA, triadin) can be detected, whereas RYR1 is depleted. [58] Nemaline rods can be detected in a minority of CCD cases. [59] Electron-microscopic features demonstrated the extensive disorganization of sarcomere in the cores, shown by a severe fragmentation and decrease of Z-bands. [60],[61] Interestingly, there is clear genotype-phenotype correlation in CCD. Mutations in the C-terminal region of RYR1 are associated with typical CCD features: characteristic, well-demarcated cores are seen, in addition to the near-absence of Type 2 fibers (in other words, almost all fibers are Type 1). In contrast, patients with mutations in the non-C-terminal region have atypical cores that are characterized with vague margin and are often multiple and peripherally-located in the muscle fiber. [40] In these patients, Type 2 fibers are variably present. There is no association between the number of cores on muscle biopsy and the degree of muscle weakness. Those with C-terminal RYR1 mutations usually have more severe symptoms with muscle weakness and scoliosis while those with non-C-terminal mutations rarely have muscle weakness although all patients with non-C-terminal mutations so far examined had MHS in our series. [40] Congenital Neuromuscular Disease with Uniform Type 1 Fibers Congenital neuromuscular disease with uniform Type 1 fibers (CNMDU1) is defined as a congenital myopathy characterized by the almost exclusive presence of Type 1 muscle fiber (>99%) without any other striking pathological changes. It was first described in 1983, [62] since then, 12 reports have been published. In our laboratory, we have 17 documented cases of CNMDU1. As discussed above, CCD due to C-terminal mutations is accompanied by the absence of Type 2 fibers [Figure 4], which is uniform Type 1 fiber, in other words, regardless of the presence of central cores, making RYR1 a potential causative gene. Indeed, sequencing of the RYR1 gene in cases with CNMDU1 revealed C-terminal mutations in four of 10 patients. [63] In fact, the clinical manifestations of the four CNMDU1 patients with C-terminal mutations are similar to those of the CCD cases due to the C-terminal mutations, regardless of the presence or absence of cores on muscle pathology, raising a possibility that they are actually the same disease, and that the absence of cores may be attributed to muscle sampling, as the degree of affectation could be different among muscles. Alternatively, cores may be formed in an age-dependent manner and in CNMDU1 cases, muscle was biopsied before core was formed. Nevertheless, in the other six cases more severe symptoms were commonly seen, such as poor fetal movement, respiratory distress and mental retardation, suggesting the involvement of the nervous system as well. Such a rare case was first reported in 1991 [64] but to date the causative gene(s) has not been discovered. Treatment and Perspectives No curative therapy is currently available for congenital myopathies. The management is essentially supportive and/or rehabilitative, based on a multidisciplinary approach. Physiotherapy is utilized for the maximum preservation of muscle power and function and the prevention of contractures, with particular care after surgical procedures. b-agonists have been shown to be beneficial on muscle strength in healthy individuals [65] and also in CCD and MmD patients. [66] This may be due to the b-adrenergic mediated fast-to-slow fiber type conversion with subsequent hypertrophy and/or reduction in wasting, however this applies only for non-progressive diseases. [67] Recent advances in identifying causative genes of the congenital myopathies, better understanding the pathomechanism, and rapidly developing diagnostic methods can provide prenatal diagnosis for these entities, although often only on a research basis. Ultimately, rational molecular therapies may be invented in the future, as most of the muscle diseases are still awaiting specific and effective treatment. References
Copyright 2008 - Neurology India The following images related to this document are available:Photo images[ni08083f3.jpg] [ni08083f4.jpg] [ni08083f2.jpg] [ni08083f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}