
|
Neurology India
Medknow Publications on behalf of the Neurological Society of India
ISSN: 0028-3886 EISSN: 1998-4022
Vol. 56, Num. 3, 2008, pp. 339-347
|
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 339-347
Review Article
Interpretation of electroneuromyographic studies in diseases of neuromuscular junction and myopathies
Mansukhani Khushnuma A, Doshi Bhavna H
Department of Clinical Neurophysiology, Bombay Hospital and Institute of Post Graduate Medicine, Mumbai and Department of Neurology, Seth G.S. Medical College and K.E.M. Hospital, Mumbai
Correspondence Address:EMG Department, Bombay Hospital, 12 Marine Lines, Mumbai - 400 020
kilom@hotmail.com
Date of Acceptance: 30-Sep-2008
Code Number: ni08085
Abstract Electroneuromyography (ENMG) also called electrodiagnosis or at times simply (and erroneously) electromyography (EMG), has been used for the diagnosis of neuromuscular and muscle diseases. It consists of tests done serially and assessed collectively, to arrive at a diagnosis of neuromuscular weakness. The test should be treated as an extension of the clinical examination and not a replacement. When done adequately ENMG is the only test which gives information about the function of the peripheral nervous system and hence co-relates well with the clinical signs. This article reviews the role of ENMG in the evaluation of neuromuscular and muscle disease. We hope this will meet the requirements of both practicing neurologists and dedicated electromyographers.
Keywords: Electromyography, myopathies, neuromuscular
Introduction
Electroneuromyography (ENMG) is the only test, or series of tests, that evaluates the function of the peripheral nervous system. It objectively documents the site of the lesion along the lower motor and sensory neuron. When assessing the peripheral nerve, it gives information about the severity, pathophysiology, extent, temporal profile, distribution and type of fiber (large or small) involvement. Serial studies help to evaluate response to treatment. When used for the diagnosis of muscle and neuromuscular diseases, it localizes the site of lesion, severity (at times), pathology, distribution, response to therapy and rules out or confirms other associated lesions. The needle electromyography examination may also indicate the most adequate muscle for biopsy and help to detect asymptomatic carriers. Though the ENMG does not predict the etiology, it can indicate or at least narrow down the differential diagnosis. However, the mainstay of the procedure lies in accurate localization of the presenting neuromuscular deficit along the peripheral nervous system.
The ENMG examination begins with a detailed history, followed by a clinical examination. This step could be shortened for the examining electromyographer by a note from the referring doctor stating what he expects from the study. The electromyographer first plans out the tests required in a particular situation, depending upon the clinical question. This is followed by the procedure itself. After the data is obtained, it is interpreted and correlated with the clinical signs and symptoms to generate the final report. Interpreting the report would hence require a clear clinical question, tailored electroneuromyographic examination, and knowledge regarding the various neuromuscular diseases.
The routine electroneuromyographic examination for the evaluation of neuromuscular disease consists of a battery of tests done serially and interpreted collectively. The nerve conduction study is generally done first and includes recording of the Sensory Nerve Action Potential from one or two relevant nerves. This is followed by Motor Nerve Conduction studies and needle electromyography (EMG) and when required, special tests are done for neuromuscular transmission defects. This review will not detail the various techniques for which excellent textbooks are available. [1],[2],[3],[4] A note of caution here would be adequate. Nerve conduction studies can be deceptively simple and EMG is still a relatively subjective analysis of the motor unit action potentials, hence great importance must be placed on the technique and experience of the physician doing the study. Interpretation of an incomplete or technically inadequate/inaccurate study would lead the physician to perform unwarranted investigations and repeat studies. The pitfalls in doing the study are therefore, equally important and clinicians must be aware of them. [5]
Test protocol The tests done for neuromuscular and muscle diseases are:
- Sensory nerve conductions
- Motor nerve Conductions
- Repetitive Nerve Stimulation: Slow (2-5 Hz) and Rapid(>5 Hz)
- Short exercise test
- Routine and Quantitative Needle EMG
- Single fiber EMG: Voluntary and Stimulated
Sensory nerve conduction
This test records the sensory nerve action potential (SNAP or SAP) which is a measure of the conduction in the sensory fibers. It gives information regarding the integrity of the peripheral nerves and hence is important for localizing the site of involvement along the peripheral nerve. The relevant nerves must be studied in a given situation and sometimes on both sides, e.g. when there is a unilateral weakness of the knee extensors (L 34 ) the Saphenous (L 34 ) SNAP should be studied as well. The SNAP is generally recorded using surface electrodes, stimulating the cutaneous nerve at one site and recording from another with a minimum distance between the two. The onset latency of the potential is a measure of the time taken for the potential to travel the distance across the two sites and is a measure of the conduction along the nerve. The amplitude of the potential, measured baseline to peak, or peak to peak is a measure of the number of fibers conducting the response. When the SNAP is abnormal (low amplitude or absent) it indicates a peripheral neuropathy.
In muscle and neuromuscular junction diseases the SNAP should be normal except when there is associated disease e.g. diabetic peripheral neuropathy. Certain muscle diseases are known to be associated with peripheral neuropathies like mitochondrial myopathies, colchicine myoneuropathy, amyloidosis, vasculitic and myotonic muscular dystrophies. Hence sensory abnormalities on nerve conduction studies along with electromyographic evidence of a muscle disease would further narrow down the differential diagnosis.
Motor nerve conduction
The motor nerve is stimulated supramaximally at two sites along its course and the response is recorded over the muscle supplied by it .This is called the compound muscle action potential and is a summation of all the individual muscle fiber action potentials. The parameters recorded are the onset latency, amplitude, duration of the response from both sites of stimulation. The conduction velocity calculated between the two sites (distance divided by the time difference between the two sites) is a measure of the conduction in the fastest conducting fibers. The onset latency of the distal response measures the conduction in the distal segment, the duration of the response is a measure of the synchrony of the conducting axons and the amplitude is an indirect measure of the number of axons conducting the response along the nerve and the number of muscle fibers responding to the stimulus. Hence the amplitude of the motor response indicates the efficacy of not only the motor nerve, but also of the neuromuscular conduction and the number of functioning muscle fibers so it is the important parameter in the evaluation of neuromuscular and muscle diseases. [6]
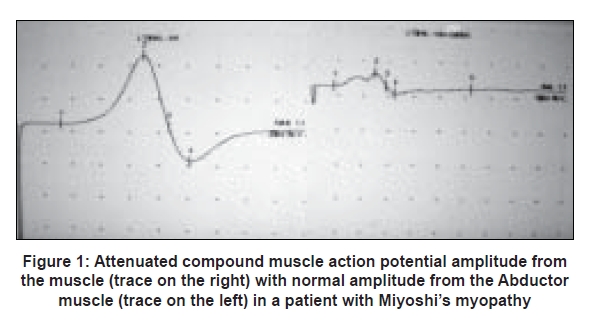
- In muscle diseases the compound muscle action potential is generally normal but can be of low amplitude if significant numbers of muscle fibers have been involved.(e.g. critical illness myopathy and end stage or severe disease)[Figure 1]. Hence amplitude from clinically severely weak muscles may be low. Compound muscle action potential amplitude increases with clinical improvement
- In pre-synaptic neuromuscular transmission defect diseases, (e.g. Lambert Eaton myasthenic syndrome) the baseline compound muscle action potential is always low amplitude as the baseline end plate potential does not reach threshold due to inadequate release of the acetylcholine quanta to single stimulus. This is seen in all muscles in Lambert Eaton myasthenic syndrome and in weak muscles in botulism
- In post-synaptic neuromuscular diseases the compound muscle action potential amplitude is usually normal at rest as the acetylcholine release at the pre-synaptic terminal is adequate and the end plate potential reaches threshold to single stimulus but can be mildly attenuated in severe involvement.
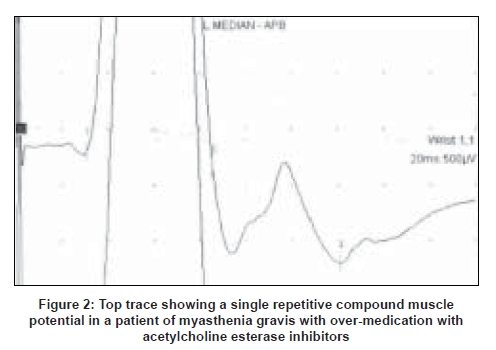
While doing the motor nerve conduction a repetitive compound muscle action potential should be looked for. The initial compound muscle action potential is immediately followed by lower amplitude single or multiple compound muscle action potentials due to accumulation of acetylcholine around the nerve terminals [Figure 2]. This is seen in congenital myasthenic syndromes e.g. slow channel syndrome, cholinesterase deficiency, over-medication with acetylcholine esterase inhibitors (AChEIs), [7],[8] organophosphate poisoning and in patients with muscle-specific tyrosine kinase (MuSK) antibodies who are receiving therapeutic doses of AChEIs. [9]
Repetitive Nerve Stimulation Test
Slow Rate Repetitive Nerve Stimulation (RNS) test : This is the most relevant test for neuromuscular transmission defects e.g. myasthenia gravis (MG), especially when abnormal (specific) but must be technically sound. [10],[11],[12] However, the sensitivity of the test is low, for ocular MG: 17% and generalized MG: 85%. [13] Also, the correlation with severity of the disease is not adequate. [14],[15] The test is more likely to yield results if multiple and proximal muscles are sampled. Normal RNS does not rule out a neuromuscular transmission defect and abnormal RNS test is very highly suggestive of MG, however, routine needle EMG of the muscle should be done to rule out anterior horn cell involvement specially if presenting symptoms are bulbar or neck muscle weakness.
The patient has to be off medication (AchE inhibitors) for 12-24 h before the test. [13] The electrodes are placed as for motor nerve conduction studies. The nerve is stimulated at one site only, recording from firmly fixed electrodes over the relevant muscle maintaining skin temperature to about 33° C. The compound muscle action potential is recorded to a single supramaximal stimulus first and the amplitude is noted. If normal, then slow rate (< 5, usually 2 per sec) repetitive stimulation is given for a train of six responses. Distal, semi-distal, proximal upper limb muscles and facial muscles are examined at rest. The test is positive when there is a decrement of at least 10% between the first and the lowest amplitude response in the train, usually the third or fourth response. [10] The decrement can be calculated using the amplitude method or ratio method. If significant decrement is observed in the muscle at rest, 10 sec voluntary exercise is given to see if the decrement reverts to normal (post exercise facilitation).[16] If there is no decrement at rest, exercise is given in the form of voluntary contraction for 1 min. The repetitive stimulus train is repeated immediately post exercise and after 2 and 4 min post exercise. Significant decrementing is looked for in the 2 min post exercise trace which tests the response of the muscle to fatigue and the recovery. [17]
Conditions in which Slow Rate Repetitive Nerve Stimulation shows decrementing responses: [11]
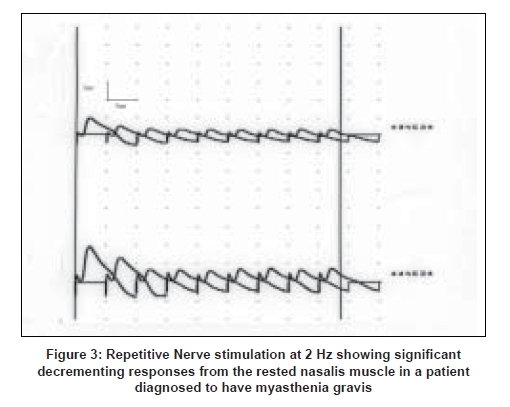
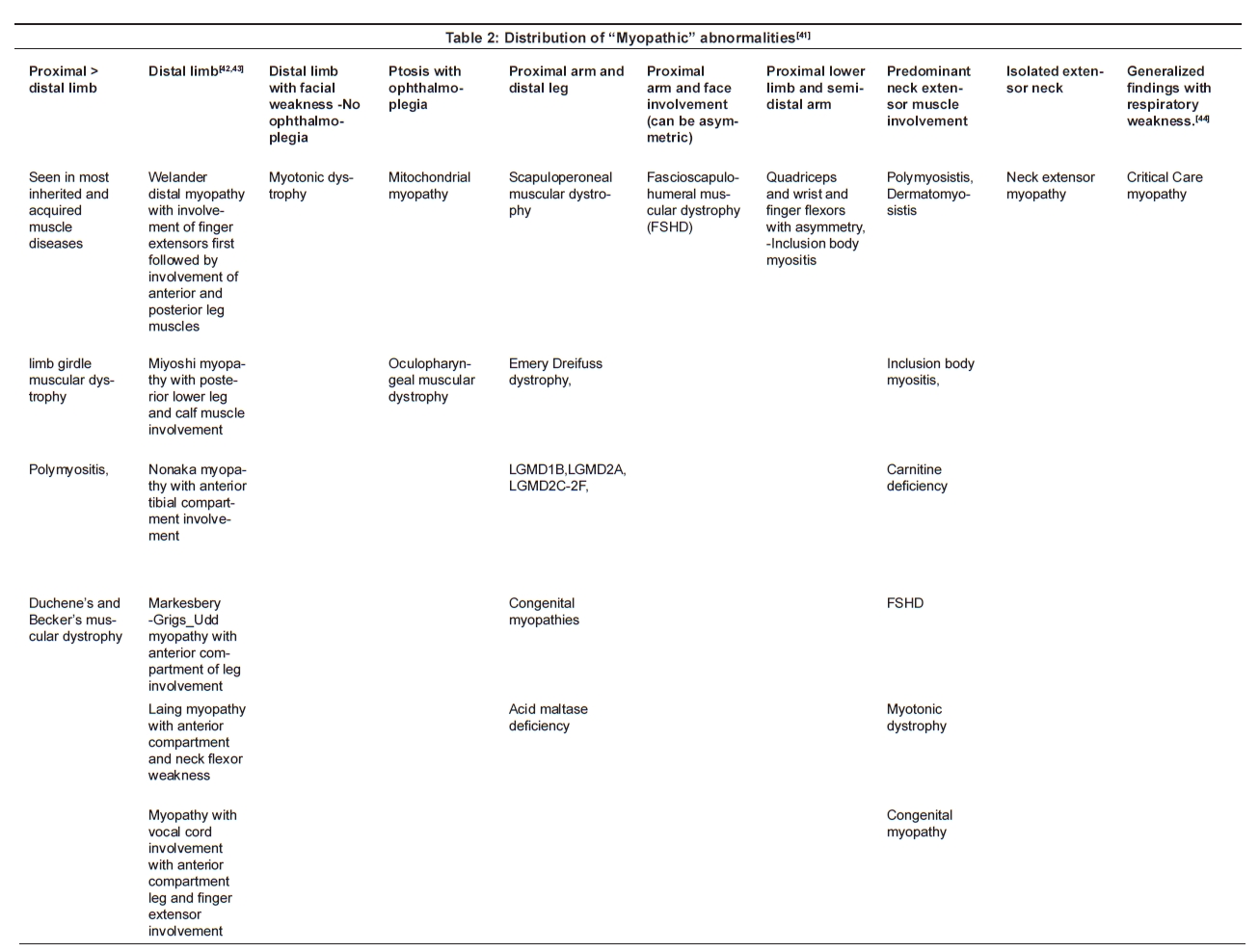
- Post-synaptic neuromuscular transmission disorders, e.g. MG: there is a significant decrement noted in the rested muscle to slow rate RNS test, most marked in the clinically weak muscle and more often in the face, neck or proximal arm muscles. [18] In 20% of patients there may be a decrement in the 2 min post exercise stimulation not noted in the rested muscle (post exercise exhaustion) [17] [Figure 3].
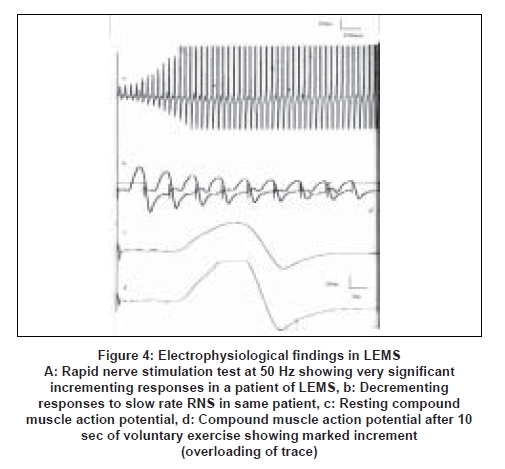
- Pre-synaptic neuromuscular diseases, e.g. Eaton Lambert Myasthenic Syndrome (LEMS), botulism [19],[20] [Figure 4] .
- Congenital myasthenic syndromes: Slow rate RNS also produces decrementing responses sometimes better demonstrated following 1 min of voluntary exercise. [7],[8]
- Paramyotonia Congenita associated with R1448C and T 1313M gene defect [21] and myasthenic syndrome caused by V1442E sodium channel mutations. [22] Slow rate RNS test demonstrates a very significant decrement, along with significant persistent post exercise fatigue.
- Amyotrophic lateral sclerosis (early stages)
- Myotonic syndromes
- Organophosphorus poisoning
Rapid rate RNS test
This is not routinely done, but may be done if a pre-synaptic neuromuscular transmission defect is suspected and there is significant incrementing response after 10 sec of exercise to a single supramaximal stimulus [Figure 4].
Rapid RNS at 25-50 Hz: There is a significant (>60%) increase [23] in the amplitude of the resting compound muscle action potential in pre-synaptic neuromuscular transmission defect, due to increased influx of calcium into the pre-synaptic terminal which results in larger release of Ach quanta.
Rapid RNS at 10-25 Hz: A few cases of autosomal recessive myotonia congenita there is a short-lasting decrement which is determined by the frequency and duration of stimulation. [24],[25]
Short exercise test
When the compound muscle action potential amplitudes are low in all muscles, with no other cause to explain them, a short voluntary exercise is given for 10 sec. [26] In pre-synaptic neuromuscular diseases: There is a very significant increase (post exercise facilitation-60-200% in Lambert Eaton syndrome and 60-100% in botulism) in the amplitude of the compound muscle action potential to a single supramaximal stimulus after the short exercise [19],[27],[28] [Figure 4]. This is due to the influx of calcium into the pre-synaptic terminal which increases the quanta and hence the baseline end plate potentials causing an increase in the compound muscle action potential amplitude.
In myotonic disorders
In paramyotonia congenita the post exercise decrement is very severe and persistent (can persist up to 40 min).Cooling of the muscle and repeated exercising increases the amount of the decrement and the time for which it lasts. [21],[25]
All paramyotonia congenita associated with T1313M and R1448C gene and one-third of the cases of myotonia congenita also show evidence of post exercise repetitive discharges (post exercise myotonic potentials) to single stimuli. [21]
A transient but significant decrement of the compound muscle action potential amplitude was noted in myotonia congenita patients with chloride channelopathies where the amplitude returned to normal within 20-40 sec after the exercise. [21]
Routine electromyography
Needle EMG is done using a concentric or monopolar needle electrode.
This is done in four steps, usually sequentially and is the most important test for primary muscle diseases.[29],[30]
- Insertional Activity
- Spontaneous Activity
- Voluntary Activity
- Interference Pattern and recruitment
The electrical activity is picked up from a muscle at i. rest, ii. With mild to moderate voluntary contraction and with iii. Full voluntary effort.
Insertional Activity : The normal muscle shows a short burst of electrical activity when the needle electrode is introduced into the muscle or moved within it, called the insertional activity(< 300 msec) When the muscle fiber membrane is irritable due to disconnection from its nerve supply due to muscle necrosis the insertional activity is increased. Myotonic discharges are a form of increased insertional activity seen in channelopathies associated with various forms of this disorder. [32] The insertional activity is reduced or may be absent with a firm feeling while inserting the needle into a fibrosed muscle or during an active attack of hypokalemic periodic paralysis.
Spontaneous Activity : The normal muscle is electrically silent at rest, except for activity near the end plate region. Spontaneous activity recorded from the muscle at rest can be fibrillations and positive sharp waves, myotonic discharges, complex repetitive discharges (site of origin being the muscle fibers), myokymic discharges, neuromyotonic discharges and fasciculation (site of origin being the terminal nerves, nerve trunk , root and anterior horn cell).
Voluntary Activity: The electrical activity picked up with mild to moderate voluntary effort is called the Motor unit Action Potential (MUP) which is a summation of activity from 8-10 muscle fibers belonging to the same motor unit and lying within the radius of the needle electrode. [33] The normal MUP has well-defined characteristics of amplitude, duration, phases and firing rate for a given muscle which get altered in diseases of the nerve, muscle and neuromuscular junction.
In myopathies the duration and amplitude of the motor unit potential is shorter than a normal unit for a given muscle as there the number of muscle fibers is reduced. [34] The motor unit also shows polyphasia as a result of desynchronized depolarization.
Interference pattern and recruitment
The firing rate and recruitment of the motor unit potential is also quantified and disease states alter that.[31] In a normal muscle with mild voluntary effort the low threshold Type II muscle fibers start firing first and the motor unit potential first recruited fires at 5-10 Hz. As the force of contraction is increased the already firing motor unit potential starts firing at faster rates and by 10 Hz the second motor unit potential starts firing (size principle). This continues as more and more units are recruited and finally individual motor unit potentials cannot be identified as each one fires at 30 Hz or more to give the pattern associated with full voluntary contraction called Interference Pattern.
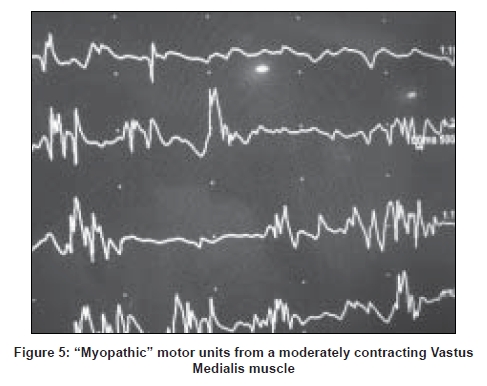
In myopathies the interference pattern is full, as motor units are normal but due to the loss of muscle fibers more units need to be recruited to produce the same voluntary effort hence the recruitment is early. That means with even a mild voluntary effort multiple short-duration low-amplitude motor units are seen. Sometimes an early recruitment may be the only abnormality. At the end stage of the disease, if too many muscle fibers are lost the recruitment would be reduced [Figure 5].
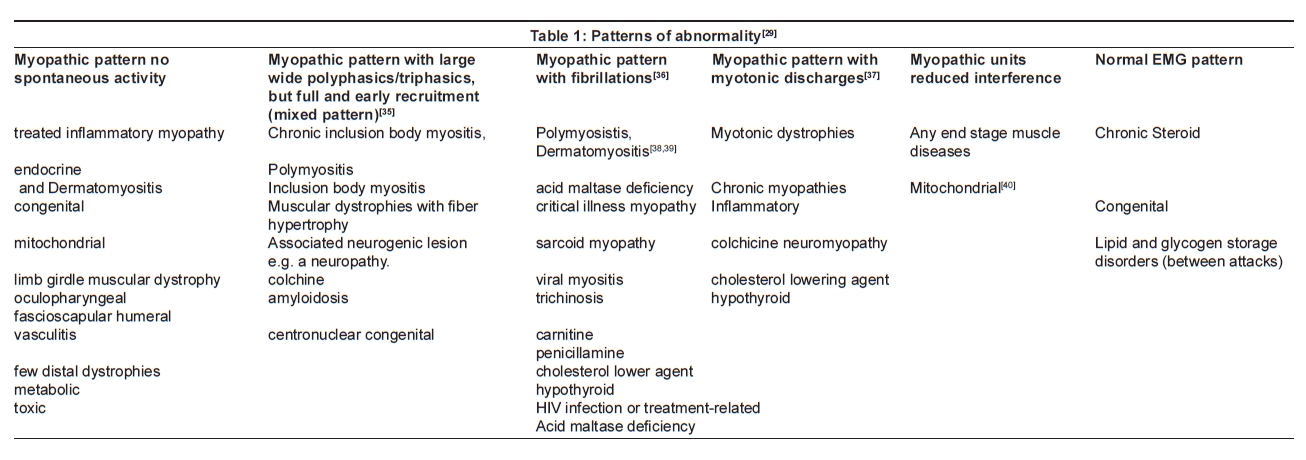
Needle EMG findings in muscle diseases [Table 1 and Table 2]
"Myopathic pattern" seen in most muscle diseases is when the motor unit action potentials recruited with mild voluntary effort show reduced dimensions (amplitude and duration) with multiple polyphasic potential and early and full recruitment.
Needle Electromyography findings in Neuromuscular Transmission disorders: [32]
Mild lesion: Routine studies are normal.
Moderate lesions: The motor units may show variation in phases and amplitude with consecutive firing, due to intermittent blocking.
Severe lesions: There is severe blocking of impulses the motor unit potentials are of short duration and low amplitude or "myopathic looking" but with normal or early interference pattern
Very severe lesions: The recruitment is also reduced along with myopathic-looking motor units.
Quantitative Electromyography
Routine needle EMG is a semi-quantitative, but predominantly qualitative analysis of the motor units and in most cases it is enough to diagnose muscle diseases. In certain cases where findings are equivocal more sophisticated computer-assisted Quantitative techniques may be required. Motor units with short rise time are isolated using a concentric needle electrode, and a steady baseline. The software program uses a fixed algorithm to analyze the units based mainly on their duration. [45],[46] Other characteristics of amplitude, phases and turns can also be quantified. [46],[47],[48] The computer analysis must be counterchecked by the electromyographer and the cursor adjustments altered when required.
Single fiber electromyography
This is a specialized study and requires expertise. [49],[50] The general principle is that instead of recording from many muscle fibers (8-10) as in conventional EMG, here a special needle electrode is used to pick up the action potentials from a smaller number of muscle fibers (two or three) innervated by the same motor unit. Hence single fiber EMG (SFEMG) is performed using a needle with a small recording surface area (25 µ m) resulting in a pickup area of 300 m 3 as compared to 1 cm 3 for regular EMG. There are two methods of picking up the single fiber potentials.
Voluntary
The single fiber needle is inserted into the muscle to be studied, (usually the Extensor Digitorum Communis, Frontalis or Orbicularis oculi) and the patient is instructed to give a mild but steady voluntary contraction. A pair of muscle potentials is recorded from a single motor unit by adjusting the needle electrode while the patient maintains the mild steady contraction. The first potential is used to trigger the screen (triggering potential) and the second muscle potential follows (slave potential) at a varying interval in relation to the first potential, called the neuromuscular jitter. Fifty consecutive discharges are recorded and the inter-potential interval is measured for each pair. Mean consecutive difference or jitter is calculated for that pair for all 50 recordings. Twenty such pairs are analyzed before a muscle is said to be normal. However, if two pairs are recorded with increased jitter the study is said to be abnormal and can be stopped there.
The second parameter measured is Neuromuscular blocking which is the intermittent failure of transmission of one of the two potentials. It is measured as a percentage of discharges that have not fired in a given pair and is abnormal only when jitter values are definitely abnormal.
Stimulated SFEMG [51]
This is useful when patient cooperation is not possible (children, comatose patients or those with movement disorders). Here the intramuscular twigs of the nerve are stimulated near the end pate zone using a monopolar electrode at fixed rate and low-intensity stimulus. The recording electrode is a single fiber electrode placed into the contracting muscle fibers. The jitter is calculated from the stimulus artifact and the single muscle fiber action potential.
Jitter analysis [11],[49],[50],[51],[52],[53] is the most sensitive test for detecting abnormalities across the neuromuscular junction provided the routine tests are all normal. Jitter values would be abnormal in myopathies and neuropathies and hence it is not a specific test in spite of its high sensitivity and must be judged in reference with other findings and clinical impression.
In our laboratory we use the following ENMG protocols. They are not watertight divisions and sometimes few more nerves need to be sampled especially if additional peripheral neuropathy is detected.
Protocol for evaluation of a patient referred for suspected muscle disease
- At least one SNAP each in the lower and upper limbs.
- Routine motor conduction study of nerves in the lower limb and upper limb.
- Record compound muscle action potential amplitude from clinically weak muscles, e.g. from Gastrocnemius in Miyoshi′s myopathy affecting the posterior Tibial muscles (here the compound muscle action potential amplitude from the Abductor Hallucis in the foot will be normal, but from the Gastrocnemius would be attenuated).
- If the compound muscle action potential amplitude is reduced in all muscles the short exercise test is done to look for post exercise facilitation. In a case of suspected critical illness myopathy, the duration of the distal compound muscle action potential is noted.
- Needle EMG examination of muscles on one side of the body, keeping the other side for muscle biopsy studies. Sample distal, semi-distal, proximal muscles of lower and upper limbs and paraspinal muscles. Face muscles when relevant and small muscles of hand if myotonic discharges are suspected are also sampled. Look for anatomic distributional pattern of abnormality e.g. hip girdle or face with proximal upper limb muscles. Sample adjacent muscles (e.g. deltoid when the trapezius and infraspinati show marked involvement)
- In children an extensive study is rarely possible, so sample the most affected muscles.
- If needle EMG is normal: Check that the most symptomatic muscles have been sampled (at least on one side). Next, a slow rate RNS test is done to rule out a neuromuscular transmission defect, before reporting it as a normal study. Follow-up studies are suggested if clinical presentation is very strongly suggestive of a muscle disease (or suggest biopsy of clinically most affected muscle and correlation with other tests).
On follow-up: Look for distribution and amount of spontaneous activity (if present earlier) and note if there is any significant increase of a previously recorded low compound muscle action potential amplitude.
Protocol for evaluation of a patient referred for suspected post-synaptic neuromuscular disease, e.g. Myasthenia Gravis
Patient should be off anticholinesterase medication for at least 12-24 h if clinically possible. If not possible, perform the test just prior to the next dose of medication.
- At least one SNAP each, in the lower and upper limb, should be normal.
- Routine motor conduction study of nerves in the lower limb and upper limb (Look for repetitive compound muscle action potentials. If baseline amplitude is low or at lower limit of normal , do the short exercise test to avoid missing a pre-synaptic defect).
- Slow rate RNS test- Sample distal hand muscle (Abductor Digiti Minimi) , Extensor Indicis, Trapezius, Nasalis, Orbicularis oculi, Massetor and Deltoid-a clinically weak muscle is more likely to yield positive results.
- If significant decrement (>10% between the first and third or fourth response) is seen in two muscles, it is positive for a post-synaptic neuromuscular transmission defect.
- If no decrement is seen in the rested muscle, voluntary exercise is given for 1 min and the RNS test repeated at 2 and 4 min intervals. If significant decrement (>10%) is seen in the trace 2 min post exercise, the test is positive.
- In restricted ocular myasthenia gravis, RNS test has a low sensitivity and that should be mentioned at the end of the report.
- Needle EMG should be performed, especially of the weak muscles (if it is a neck-bulbar presentation, with no ocular symptoms) to rule out anterior horn cell involvement and look for variation of motor unit potential configuration.
- If the slow rate RNS is not positive, SFEMG is done. If expertise is not available for this, other laboratory test should be used (antibodies to acetylcholine receptors or MuSK antibodies).
Protocol for evaluation of a patient referred for suspected pre-synaptic neuromuscular disease, e.g. Lambert Eaton Myasthenic Syndrome
- One SNAP each, in lower and upper limbs
- Routine motor conduction study of nerves in the lower limb and upper limb-compound muscle action potential amplitude should be of low amplitude in all muscles in Lambert Eaton Myasthenic Syndrome and in proximal and weak muscles in botulism.
- Short exercise test (10 sec) is done to look for post exercise facilitation, 60-200% in Lambert Eaton Myasthenic Syndrome, 60-100% in botulism.
- Slow rate RNS to demonstrate the decrement.
- Rapid nerve stimulation may be done, however, the demonstration of unequivocal post exercise facilitation to a single stimulus is enough to confirm a pre-synaptic neuromuscular transmission defect.
- Routine needle EMG to rule out anterior horn cell involvement.
- Follow-up studies on treatment -to look for improvement of the baseline amplitude at rest.
Acknowledgments Dr. Jayanti Mani: for reading the paper and offering her valuable suggestions. Dr. S.S. Pandya: my teacher and driving force.[54]
References
| 1. | Aminoff MJ. Electromyography in clinical practice. 3rd ed. New York: Churchill Livingstone; 1998. Back to cited text no. 1 |
| 2. | Oh SJ. Clinical electromyography: nerve conduction studies. 3rd ed. Baltimore: Lippincott Williams and Wilkins; 2002. Back to cited text no. 2 |
| 3. | Kimura J. Electrodiagnosis in diseases of nerve and muscle: principles and practice, 3rd ed. New York: Oxford University Press; 2001. Back to cited text no. 3 |
| 4. | Johnson EW, Pease WS. Practical electromyography. 3rd ed. Baltimore: Williams and Wilkins; 1997. Back to cited text no. 4 |
| 5. | Kimura J. Facts, fallacies, and fancies of nerve conduction studies: Twenty-first annual Edward H. Lambert Lecture. Muscle Nerve 1997;20:777-87. Back to cited text no. 5 |
| 6. | Wilbourn AJ. Nerve conduction studies in axonopathies and demyelinating neuropathies: Fundamentals of Electrodiagnosis. Rochester, MN: American Association of Electrodiagnostic Medicine; 1989. p. 7-24. Back to cited text no. 6 |
| 7. | Engle AG. The myasthenic syndromes. New York: Oxford University Press; 1999. Back to cited text no. 7 |
| 8. | Engle AG. Congenital myasthenic syndromes. In: Katirji B, Kaminski HJ, Preston DC, Ruff RL, Shapiro EB, editors. Neuromuscular disorders in clinical practice. Boston: Butterworth-Heinemann; 2002. p. 953-63. Back to cited text no. 8 |
| 9. | Punga AR, Flink R, Askmark H, Stεlberg EV. Cholinergic neuromuscular hyperactivity in patients with myasthenia Gravis seropositive for MuSK antibody. Muscle Nerve 2006;34:111-5. Back to cited text no. 9 |
| 10. | Shapiro BE, Katirji B, Preston DC. Clinical electromyography. In: Katirji B, Kaminski HJ, Ruff RL, Shapiro EB, editors. Neuromuscular disorders in clinical practice. Boston: Butterworth-Heinemann; 2002. p. 80-140. Back to cited text no. 10 |
| 11. | Oh SJ. Electromyography: Neuromuscular transmission studies. Baltimore: Williams and Wilkins; 1998. Back to cited text no. 11 |
| 12. | AANEM Practice parameter for repetitive nerve stimulation and single fibre EMG evaluation of adults with suspected myasthenia gravis or Lambert-Eaton myasthenic syndrome: summary statement. Muscle Nerve 2001;24:1236-8. Back to cited text no. 12 |
| 13. | Oh SJ, Eslami N, Nishihira T, Sarala PK, Kuba T, Elmore RS, et al . Electrophysiological and clinical correlation in myasthenia gravis. Ann Neurol 1982;12:348-54. Back to cited text no. 13 |
| 14. | Gilchrist JM, Massey JM, Sanders DB. Single fibre EMG and repetitive stimulation of same muscle in myasthenia gravis. Muscle Nerve 1994;17:171-5. Back to cited text no. 14 |
| 15. | Oey PL, Wieneke GH, Hoogenraad TU, van Huffelen AC. Ocular myasthenia gravis: The diagnostic yield of repetitive nerve stimulation and stimulated single fibre EMG of Orbicularis oculi muscle and infra red reflection oculography. Muscle Nerve 1993;16:142-9. Back to cited text no. 15 |
| 16. | Desmedt JE. The neuromuscular disorder in myasthenia gravis: Electrical and mechanical response to nerve stimulation in the hand muscles: New Developments in EMG and Clinical Neurophysiology. Vol 1. Karger: Basel; 1973. p. 241-2. Back to cited text no. 16 |
| 17. | Rubin DI, Hentschel K. Is exercise necessary with repetitive nerve stimulation in evaluating patients with suspected myasthenia gravis? Muscle Nerve 2007;35:103-6. Back to cited text no. 17 |
| 18. | Oh SJ, Kim DE, Kuruoglu R, Bradley RJ, Dwyer D. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve 1992;15:720-4. Back to cited text no. 18 |
| 19. | Keesey JC. Electrodiagnostic approach to defects of neuromuscular transmission. Muscle Nerve 1989;12:613-26. Back to cited text no. 19 |
| 20. | Fakadej AV, Guttmann L. Prolongation of post titanic facilitation in infant botulism. Muscle Nerve 1982;5:727-9. Back to cited text no. 20 |
| 21. | Fournier E, Arzel M, Sternberg D, Vicart S, Laforet P, Eymard B, et al . Electromyography guides towards subgroups of mutations in muscle channelopathies. Ann Neurol 2004;56:650-61. Back to cited text no. 21 |
| 22. | Tsujino A, Maertens C, Ohno K, Shen XM, Fukuda T, Harper CM, et al . Myasthenic syndrome caused by mutation of the SCN4A sodium channel. Proc Natl Acad Sci USA 2003;100:7377-82. Back to cited text no. 22 |
| 23. | Lambert EH, Rooke ED, Eaton LM. Myasthenic syndrome occasionally associated with bronchial neoplasm: Neurophysiological studies. In: Viets HR, editor. Myasthenia Gravis: The second International Symposium Proceedings 1959. Springfield, IL: Charles C. Thomas; 1961. p. 362-410. Back to cited text no. 23 |
| 24. | Aminoff MJ, Layzer RB, Satya-Murti S, Faden AI. The declining electrical response of muscle to repetitive nerve stimulation in myotonia. Neurology 1977;27:812-6. Back to cited text no. 24 |
| 25. | Streib EW, Sun SF, Yarkowsky T. Transient paresis in myotonic syndromes: A simplified Electrophysiologic approach. Muscle Nerve 1982;5:719-23. Back to cited text no. 25 |
| 26. | Hatanaka Y, Oh SJ. Ten second exercise is superior to 30 second exercise for post exercise facilitation in diagnosing Lambert Eaton myasthenic syndrome. Muscle Nerve 2008;37:572-5. Back to cited text no. 26 |
| 27. | Oh SJ. Botulism: Electrophysiologic studies. Ann Neurol 1977;2:481-5. Back to cited text no. 27 |
| 28. | Oh SJ, Kurokawa K, Claussen GC, Ryan HF Jr. Electrophysiological diagnostic criteria of Lambert Eaton myasthenic syndrome. Muscle Nerve 2005;32:515-20. Back to cited text no. 28 |
| 29. | Lacomis D. Electrodiagnostic approach to the patient with suspected myopathy. Neurol Clin N Am 2002;20:587-603. Back to cited text no. 29 |
| 30. | Daube JR. Needle examination in clinical electromyography. Muscle Nerve 1991;14:685-700. Back to cited text no. 30 |
| 31. | Petajan JH. Motor unit recruitment. Muscle Nerve 1991;14:489-502. Back to cited text no. 31 |
| 32. | Preston DC, Shapiro B. Needle electromyography Fundamentals, normal and abnormal patterns. Neurol Clin N Am 2002;20:361-96. Back to cited text no. 32 |
| 33. | Nandedkar SD. Objective EMG: Quantitation and documentation in the routine needle electromyographic examination. In: Johnson EW, Pease WS, editors. Practical electromyography. 3rd ed. Baltimore: Williams and Wilkins; 1997. p. 41-61. Back to cited text no. 33 |
| 34. | Buchthal F. Diagnostic significance of the myopathic EMG. In: Rowland LP, editor. Pathogenesis of human muscular dystrophies. Proceedings of the fifth international scientific conference of muscular dystrophy association. Amsterdam: Excerpta Medica; 1977. p. 205-18. Back to cited text no. 34 |
| 35. | Uncini A, Lange DJ, Lovelace RE, Solomon M, Hays AP. Long duration polyphasic motor unit potentials in myopathies: A quantitative study with pathological co-relation. Muscle Nerve 1990;13:263-7. Back to cited text no. 35 |
| 36. | Lacomis D, Giuliani Mj, Van Cott A, Kramer DJ. Acute myopathy on intensive care: Clinical, electromyographic and pathological aspects. Ann Neurol 1996;40:645-54. Back to cited text no. 36 |
| 37. | Streib EW. Differential diagnosis of myotonic syndromes. Muscle Nerve 1987;10:603-15. Back to cited text no. 37 |
| 38. | Amato AA, Barohn RJ. Idiopathic inflammatory myopathies. Neurol Clin 1997;15:615-48. Back to cited text no. 38 |
| 39. | Dalakas MC, Hohlfeld R, Polymyosistis and dermatomyositis. Lancet 2003;362:971-82. Back to cited text no. 39 |
| 40. | Petty RK, Harding AE, Morgan-Hughes JA. The clinical features of mitochondrial myopathy. Brain 1986;109:915-38. Back to cited text no. 40 |
| 41. | Barohn RJ. General approach to muscle diseases In: Goldman L, Ausiello D, editors. Cecil textbook of medicine. 22nd ed. Philadelphia: WB Saunders; 2004. p. 2370-9. Back to cited text no. 41 |
| 42. | Saperstein DS, Amato AA, Barohn RJ. Clinical and genetic aspects of distal myopathies. Muscle Nerve 2001;24:1440-50. Back to cited text no. 42 |
| 43. | Udd B, Griggs R. Distal myopathies. Curr Opin Neurol 2001;14:561-6. Back to cited text no. 43 |
| 44. | Bolton CF. Neuromuscular manifestations of critical illness. Muscle Nerve 2005;32:140-63. Back to cited text no. 44 |
| 45. | Nandedkar SD, Barkhaus PE, Charles A. Multi-motor unit action potential analysis (MMA). Muscle Nerve 1995;18:1155-66. Back to cited text no. 45 |
| 46. | Dorfman LJ, McGill KC. Automatic quantitative electromyography. Muscle Nerve 1988;11:804-18. Back to cited text no. 46 |
| 47. | Willison RG. Analysis of electrical activity in healthy and dystrophic muscle in man. J Neurol Neurosurg Psychiatry 1964;27:386-94. Back to cited text no. 47 |
| 48. | Stεlberg E, Andreassen S, Falck B, Lang H, Rosenfalck A, Trojaborg W. Quantitative analysis of individual motor unit action potentials: A proposal for standardized terminology and criterion for measurement. J Clin Neurophysiol 1986;3:313-48. Back to cited text no. 48 |
| 49. | Stεlberg E, Trontelj JV. Single fibre electromyography in healthy and diseased muscles. New York: Raven Press; 1994. Back to cited text no. 49 |
| 50. | Stεlberg E, Sanders DB. Electrophysiological tests of neuromuscular transmission. In: Stεlberg E, Young R, editors. Clinical Neurophysiology. London: Butterworths; 1981. Back to cited text no. 50 |
| 51. | Trontelj JV, Mihelin M, Fernandez JM, Stεlberg E. Axonal stimulation for end plate jitter studies. J Neurol Neurosurger Psychiatry 1986;49:677-85. Back to cited text no. 51 |
| 52. | Stεlberg E, Ekstedt J, Broman A. The electromyographic jitter in human muscles: Electroencephalgr Clin Neurophysiol. 1971;31:429-38. Back to cited text no. 52 |
| 53. | Sanders DB, Howard JF Jr. Single fibre electromyography in myasthenia gravis. Muscle Nerve 1986;9:809-19. Back to cited text no. 53 |
| 54. | Sanders DB, Howard JF Jr, Johns TR. Single fibre EMG in myasthenia gravis. Neurology 1979;29:68-76. Back to cited text no. 54 |
Copyright 2008 - Neurology India
The following images related to this document are available:
Photo images
[ni08085t2.jpg]
[ni08085t1.jpg]
[ni08085f5.jpg]
[ni08085f2.jpg]
[ni08085f4.jpg]
[ni08085f3.jpg]
[ni08085f1.jpg]
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}