 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
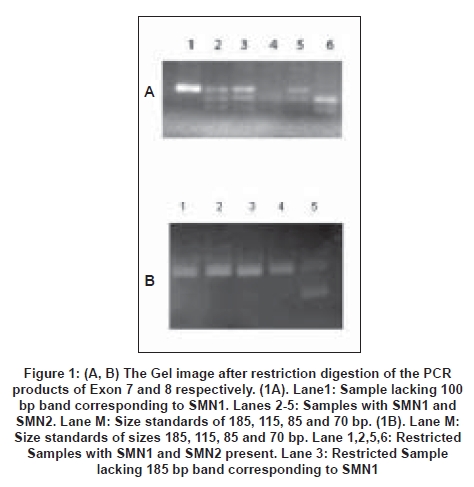
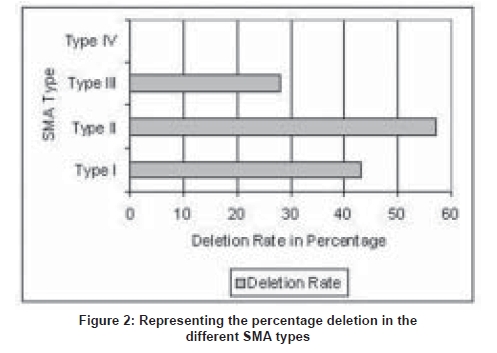
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 348-351 Original Article Deletion analysis of spinal muscular atrophy in southern Indian population Swaminathan Bhairavi, Shylashree S, Purushottam Meera, Taly AB, Nalini A Molecular Genetics Laboratory, Department of Neurology, National Institute of Mental Health and Neurosciences, Bangalore Date of Acceptance: 05-Sep-2008 Code Number: ni08086 Abstract Background: Proximal spinal muscular atrophy (SMA) is a genetically heterogeneous disease with paresis and muscle atrophy due to loss of anterior horn cell function. The survival of motor neuron gene (SMN) and neuronal apoptosis inhibitory protein (NAIP) play a primary role. Both the gene homologues exist as inverted duplications on Chromosome 5q. The telomeric/functional (SMN1) and the centromeric (SMN2) copies differ from each other in eight nucleotides. The C→T transition (at Codon 280) within Exon 7 of SMN2 causes disruption of an exonic splicing enhancer (ESE) and/or creates an exonic splicing silencer (ESS) leading to abnormal splicing and a truncated protein.Objective: To determine the molecular genetics of SMN1 and NAIP genes in SMA from southern India. Materials and Methods: In the present study, 37 patients from the neuromuscular disorders clinic of National Institute of Mental Health and Neurosciences were assayed for the deletions in the SMN1 and NAIP genes using PCR-RFLP methods. Results: Among the SMA Type I patients, 43% showed deletions of SMN1 and NAIP. In patients Type II SMA, 57% showed deletions of the SMN1 exons. Conclusion: Thus, deletions were found to occur in 47.8% of the Type I and II patients. Lower sensitivity of gene deletion study in clinically suspected SMA needs further study as clinical diagnosis of SMA is not gold standard. However, the results do correlate with other studies conducted in India. Keywords: Genetics, southern India, spinal muscular atrophy Introduction Proximal spinal muscular atrophy (SMA) is a genetically heterogeneous disease with paresis and muscle atrophy due to loss of anterior horn cell function. [1] The disease varies from onset in prenatal period to adulthood. [2] This neuromuscular disorder is the second most common and lethal disorder with an incidence rate between 1 in 6000 to 1 in 10,000 live births, and a carrier frequency of 1 in 40 to 1 in 60. [3],[4],[5],[6] SMA is clinically divided into several subgroups, (Types I-IV) based on the age of onset and motor disabilities. [7] SMA Type I (acute SMA; MIM 253300) also known as Werdnig-Hoffman disease has the most severe phenotype. [8] Onset of clinical symptoms in SMA Type I is by definition before the age of six months and progressive muscle weakness and subsequent leads to rapid progressive course in most and death before the age of two years. [9] Children with Type II SMA (intermediate form; MIM 253550) show onset after six months. They can sit but are never able to stand or walk, unaided, and life expectancy is significantly reduced. In SMA Type III (Kugelberg-Welander disease; MIM 253400) there is a broader range of age at onset, patients with an age of onset before three years are classified as Type IIIa and those with onset after three years as Type IIIb. [10] Such children are able to stand and walk unaided; however they become wheelchair-bound in adolescence or adulthood. Patients with an age of onset beyond 30 years are classified as Type IV SMA. SMA can be confused with clinically overlapping neurological disorders [1] and the subdivision between acute and chronic SMA is nearly arbitrary, with overlap between the groups . [1] In 1990 the gene locus for Type I-III SMA was mapped by linkage studies to Chromosome 5q13. [1],[11],[12],[13] Chronic childhood onset SMA (SMAI, II and III) maps to a single locus of Chromosome 5q11.2-13.3. [13] Among the candidate genes located in this region, the survival of motor neuron (SMN) gene and the neuronal apoptosis inhibitory (NAIP ) gene were suggested to play an important role. [13],[14],[15],[16],[17] The SMN and the NAIP genes are found as two homologous copies, existing as inverted duplications. Both the copies of SMN genes contain nine exons, and differ from each other in eight nucleotides and can be differentiated by the use of restriction enzymes. The sixth nucleotide of Exon 7 in SMN2 contains a T nucleotide, which disrupts an exonic splicing enhancer and/or creates an exonic splicing silencer (ESS), leading to alternative splicing, resulting in skipping of Exon 7 in around 90% of the SMN2 transcripts. These transcripts lead to the formation of an unstable and truncated protein that is degraded, thus making the SMN1 the functional copy. In patients with SMA, SMN1 is found to be deleted in a homozygous fashion, with the SMN2 failing to compensate for the loss of SMN1 . [18],[19] Similar such reports have been published from different parts of India. [20],[21] Materials and Methods Clinical Genetics SMN Gene: The PCR amplification of SMN Exon 7 with Reverse Primer: 5′-CTACAACACCCTTCTCACAG-3′) were done as per the protocol of Wirth et al . [23] Both Exon 7 and 8 were amplified using the following PCR conditions: initial denaturation 95 o C for 5 min; followed by 34 cycles of 95 o C for 30 sec; 55 o C for 30 sec; 72 o C for 30 sec; final extension of 72 o C for 7 min.The 135 bp amplified product of Exon 7 was digested with HinfI , and run on 12% PAGE . The presence of particular size bands allowed differentiation of SMN2 ( SMN2 -100 bp, 35 bp) from SMN1 ( SMN1 -80 bp, 20 bp, 35 bp) [Figure 1A]. The 185 bp product of Exon 8 was digested with DdeI , and run on a 2% agarose gel. An undigested PCR product was indicative of the functional SMN1 [Figure 1B]. In a few patient samples a band of lighter intensity was obtained after digestion compared to the control indicative of a heterozygous state and was confirmed by sequencing of the PCR product. NAIP Gene: Amplification of Exon 5 (Forward Primer: 5′-AAGCCTCTGACGAGAGGATC-3′; Reverse Primer 5′-CTCTCAGCCTGCTCTTCAGAT-3′) NAIP gene was carried out using the following PCR conditions; initial denaturation 94 o C for 10 min; followed by 30 cycles of 94 o C for min , 60 o C for 30 sec, 72 o C for 30 sec; final extension at 72 o C for min . Exon deletions were indicated by the absence of a 435 bp band. Results The mean age of onset for the childhood onset SMA (I, II and III) was 1.76 ± 1.93 years (0.08-7.00) at the time of evaluation. Thirteen of the affected children were born to consanguineous parents (uncle-niece (21.6%), first cousin (13.5%)). Of these nine patients (males-five; females-four) had affected sibling(s). The creatinine kinase level among SMA Type I ranged from 37-678 U/L (mean- 264), SMA Type II, 250 - 462 U/L (mean -384), SMA Type III, 64 - 356 U/L (mean 135), SMA Type IV 109 - 920 U/L (mean - 460). Although the levels were above normal limits in the majority, they were not remarkably elevated. Muscle sample for histopathological study was obtained from the biceps in 14 patients belonging to SMA Type II-IV. The findings were suggestive of neurogenic process with evidence of atrophy and hypertrophic fibers and group atrophy. Among the 16 patients clinically diagnosed to have SMA Type I, seven (43%) showed deletions in the SMN and/or NAIP genes [Figure 2]. The age of onset varied from third trimester to seven months. Positive muscle biopsy could be obtained in three of these individuals. In seven patients with clinical features of SMA Type II, four (57%) patients showed deletion of the SMN and/or NAIP genes [Figure 2]. The age of onset ranged from one to two years. Three showed neurogenic process in muscle biopsy. In seven patients clinically diagnosed to have SMA Type III patients, two (28%) patients showed deletion of SMN and/or NAIP genes [Figure 2]. The age of onset ranged from three to seven years. Of the seven SMA Type IV patients, none showed deletions of the SMN exons [Figure 2]. Discussion SMA was first recognized as a distinct entity about 115 years ago, with the nearly simultaneous clinicopathologic descriptions of Werdnig and Hoffman. [24],[25],[26] SMA is one of the most frequent serious genetic disorders in children with an estimated birth prevalence of 1 in 10,000 births and a carrier frequency of 1 in 45 individuals. [27] In our study, SMA Type I was uniformly defined by an onset before six months with severe weakness; however by genetic analysis only 43% showed the deletion. Among the SMA Type II cases a higher number of patients demonstrated the deletion. Classification on the basis of clinical findings does not always permit unambiguous sub-grouping since the disease has phenotypic similarities with other childhood/floppy infant syndromes. [12] Molecular diagnosis of childhood proximal SMA has been possible after the discovery of the SMN gene. [3],[18] Deletions of Exons 7 and 8 of the SMN gene occur in 98.6% and 93.0% of patients respectively, independent of the disease severity. [28] NAIP is disrupted in 45% of patients with Type I disease and in 18% of SMA II and III individuals and is thought to contribute to the severity of this disorder. [29] The study by Labrum et al. , reported a SMN1 deletion rate of 51% among the South African SMA population. [30] In a recent study by Dastur et al. , from India, a deletion rate for SMN1 of around 73% in Type I and 26% in Type II SMA has been reported. [31] Dua et al ., in a study from North India reported deletions of the SMN1 gene in 50% of cases. [32] In another study by Kesari et al. , from northern India, a homozygous deletion rate of 76% in SMA patients was demonstrated. [20] In our study, the total rate of deletions in SMA Type I and II patients, accounted for around 47.8%. Biopsies were done in 14 patients which showed neurogenic process. Among the 16 SMA Type I patients, eight showed neurogenic process on muscle biopsy and three of these demonstrated deletions in the SMN and/or NAIP genes. Of the four Type II patients with neurogenic process on muscle biopsy, three of them showed deletions of the SMN and/or NAIP genes. In a study with adult onset SMA, the SMN gene was found to be deleted in all patients. These authors propose that there is genetic homogeneity between the clinically diverse adult onset form and childhood forms of the disease. [33] However, in our study the seven patients with adult onset SMA showed no deletions of the SMN or NAIP genes. An attempt was made to correlate the pathological and genetic findings. However, it did not demonstrate a direct correlation in all; this could be due to mutations or gene conversions that are being reported to cause SMA. These iatrogenic mutations and gene conversions could also be a possible explanation for the low rates of detected deletions in our study. It is also possible that particularly among the SMA Type I patients there could have been non-SMA floppy infant cases included as clinical assessment was utilized in the majority. The current diagnostic method of SMA is predominantly by the use of invasive techniques like muscle biopsy which is not a preferred method of diagnosis for infants. The advantage of using the PCR method for diagnostics is that it is convenient and noninvasive and could be preferred in infants and young children. Further, this method of genetic testing can be used in conjunction with other markers for prenatal diagnosis. Acknowledgment The authors acknowledge the Indian Council of Medical Research (ICMR), New Delhi for financial support extended to the ′Central Molecular Genetics Laboratory for Neurology and Psychiatry′.References
Copyright 2008 - Neurology India The following images related to this document are available:Photo images[ni08086f2.jpg] [ni08086f1.jpg] |
| |||||||||


{kind=link}
{kind=link}