 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
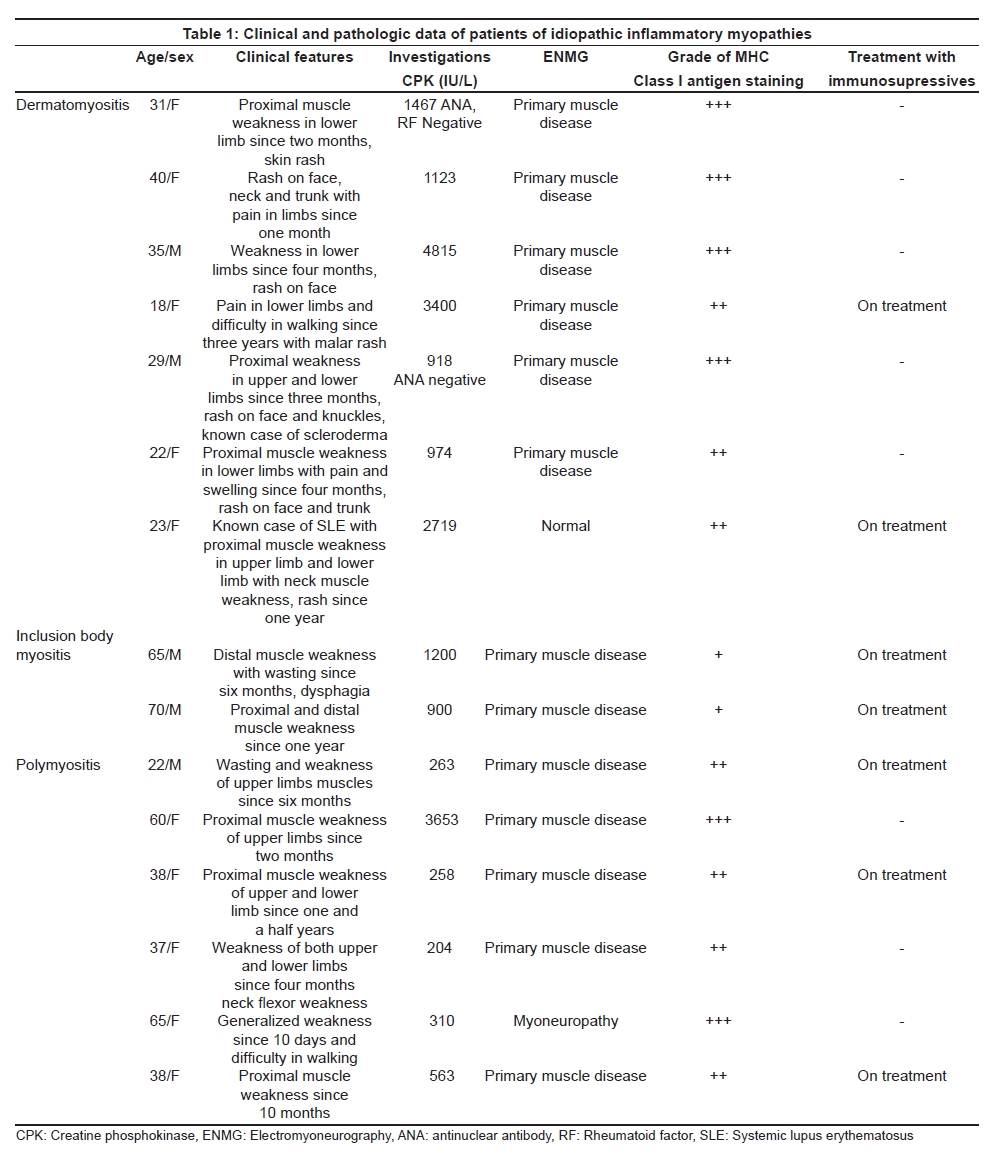
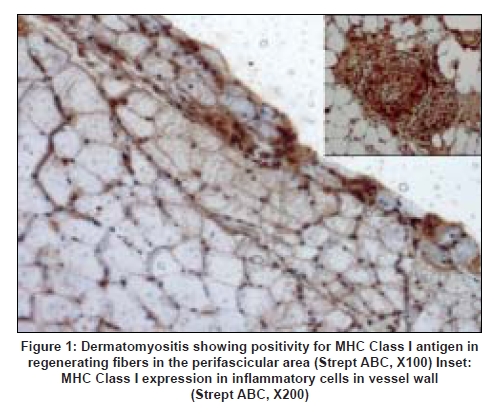
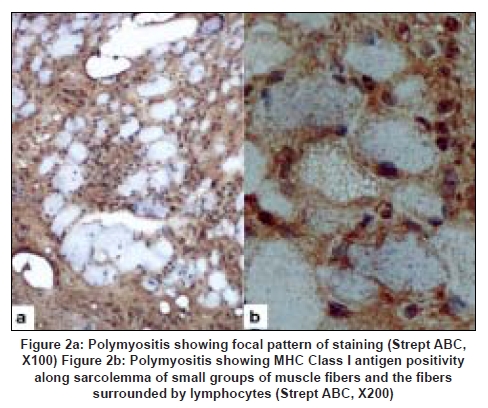
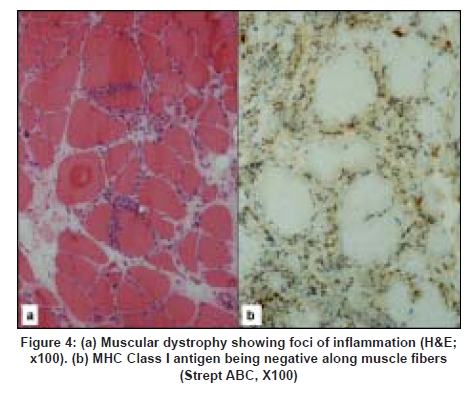
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 363-367 Original Article Major histocompatibility complex class I expression can be used as a diagnostic tool to differentiate idiopathic inflammatory myopathies from dystrophies Sundaram C, Uppin Megha S, Meena AK Department of Pathology, Nizam's Institute of Medical Sciences, Punjagutta, Hyderabad Date of Acceptance: 12-Jun-2008 Code Number: ni08089 Abstract Aim: Utility of major histocompatibility complex (MHC) Class I antigen immunostaining was studied to differentiate idiopathic inflammatory myopathies from dystrophies.Materials and Methods: Forty muscle biopsies including seven dermatomyositis (DM), six polymyositis (PM), two sporadic inclusion body myositis (sIBM), 20 dystrophies (one Duchenne, three Becker's, four alpha, one gamma sarcoglycanopathy, nine limb girdle, one myotonic and one fascioscapulohumeral muscular dystrophy) and five controls were stained with antibody for MHC Class I antigen (Novocastra clone W6/32 HL 1:100 dilution). Results: Polymyositis and sIBM showed MHC class I antigen positivity along sarcolemma of single and small groups of muscle fibers. The regenerating fibers in the perifascicular area in DM showed intense cytoplasmic positivity of MHC class I antigen. Muscle fibers in all dystrophies except regenerating fibers and control normal muscle were negative for MHC. Capillaries and lymphocytes were positive controls. There were no false positives in the study. Conclusion: MHC Class I immunostaining can be used as a complementary diagnostic tool for the diagnosis of idiopathic inflammatory myopathies. Keywords: Dermatomyositis, inclusion body myositis, inflammatory myopathies, major histocompatibility complex, muscular dystrophy, polymyositis Introduction Idiopathic inflammatory myopathies (IIM) are a heterogeneous group of disorders and include dermatomyositis (DM), polymyositis (PM) and sporadic inclusion body myositis (sIBM). These are clinically important because they are potentially treatable. Hence accurate diagnosis is important for treatment and prognosis. Presence of mononuclear cells in the muscle biopsy is essential for the diagnosis according to the criteria by Bohan and Peter. [1],[2] However, inflammatory cells can occur in toxic, necrotizing and dystrophic muscle diseases, especially fascioscapulohumeral muscular dystrophy (FSHD), Duchenne muscular dystrophy (DMD), dysferlinopathy and other limb girdle muscular dystrophies (LGMD), leading to misdiagnosis. Sampling error, inadequate processing techniques, misinterpretation and treatment instituted before biopsy can constitute wrong diagnosis and inflammatory cells may be absent even in the presence of disease. Hence there is a need for a better and valid test to diagnose IIM and categorize them into DM, PM and sIBM and also to differentiate them from other muscle diseases. Major histocompatibility complex (MHC) Class I antigen is absent in normal muscle fibers and is upregulated in IIM. [3] Immunohistochemical detection of MHC Class I antigen was used as a diagnostic test for IIM by van der Pas et al . [4],[5] So we aim to study the utility of MHC Class I antigen expression for the diagnosis of IIM and to differentiate them from other muscle diseases. Materials and Methods It was a retrospective study performed on 40 muscle biopsies including 15 cases of IIM, 20 cases of dystrophies and five normal muscles during the period January 2005 to June 2006. All biopsies included in the study except controls showed foci of inflammation including IIM. All patients with IIM included in the study were diagnosed by criteria of Bohan and Peter. [1] This included seven patients of DM, six patients of PM and two patients of sIBM. The dystrophies included one DMD, five sarcolglycanopathy (four α and one γ sarcoglycanopathies), nine LGMD unclassified, three Becker′s muscular dystrophy (BMD), one fascioscapulohumeral dystrophy (FSHD) and one myotonic dystrophy. The dystrophies were categorized based on clinical picture, family history and immunohistochemistry using dystrophin 1, 2, 3, sarcoglycans α, β, γ, δ (Novocastra antibodies). In all the patients, muscle biopsy was done from the left vastus lateralis by open biopsy and the sample was divided into three parts. One part was immediately fixed in buffered gluteraldehyde for electron microscopy. One part was snap-frozen in isopentane precooled in liquid nitrogen and transferred to cryostat and sections were cut. The third part was fixed in buffered formalin and processed for paraffin sections. Cryosections were stained with Hematoxylin and Eosin, Masson trichrome, modified Gomori′s trichrome (MGT), adenosine triphosphatase (ATPase) preincubated at pH 9.4, 4.6 and 4.3, succinic dehydrogenase (SDH), nicotinamide adenine dinucleotide tetrazolium reducatse (NADH-TR). Immunohistochemistry was performed on cryosections using monoclonal antibodies to MHC Class I by streptavidin-biotin technique. Five-micron thickness cryostat sections were first brought to room temperature and fixed in acetone methanol. Protein block was applied for 5 min. Further the sections were incubated with primary antibody (Novocastra HLA-ABC, clone W6/32-HL Novocastra Ltd., Newcastle Upon tyne, UK) for one hour in 1:100 dilution. This was followed by incubation with secondary antibody for 30 min and further incubation with streptavidine-HRP. Peroxidase activity was developed by diaminobenezidine. The slides were counterstained with Hematoxylin, dehydrated and mounted in DPX Di butyl pthalate xylene. Capillaries in normal muscle and lymphocytes served as internal positive controls and omission of primary antibody served as negative control. The results of immunohistochemistry were independently evaluated by two pathologists (C. S and U. M) and consensus was taken. The intensity of staining was graded from 0 to +++ as follows. 0: No staining, +: Weak, ++: Moderate intensity, +++: Strong intensity. There was no correlation of intensity of staining to inflammation or regenerative activity. Results Table 1 gives the clinical, pathological and therapeutic details of all patients with IIM. Dermatomyositis (n=7): The age ranged from 22 to 40 years with a mean of 24 years. There were two males and five females. All patients presented with skin rash and proximal muscle weakness. Two patients had associated collagen vascular disease. Creatine phosphokinase (CPK) was elevated in all patients. Antibody levels were assessed in three patients of which one patient showed negative antinuclear antibody(ANA) and rheumatoid factor and the other patient of systemic lupus erythematosus was positive for anti ds DNA antibody. The patient of scleroderma tested negative for ANA. Two patients were already started on immunosuppressive therapy. Muscle biopsy showed characteristic perifascicular atrophy, regenerating fibers and endomysial and perivascular inflammation. The regenerating fibers in the perifascicular area showed intense cytoplasmic positivity for MHC Class I antigen [Figure 1]. The capillaries and lymphocytes showed positivity (Figure 1 inset) and muscle fibers were negative. Polymyositis (n=6): The age ranged from 20-45 years with a mean of 27.6 years. There was one male and five female patients. All of them presented with proximal muscle weakness. There was no associated collagen vascular disease. The CPK was elevated in all patients. Three of these patients were on immunosuppressive therapy. Muscle biopsy showed inflammation around non-necrotic fibers. There were no regenerating fibers. The MHC Class I antigen was expressed along sarcolemma of small groups of non necrotic muscle fibers and fibers surrounded by lymphocytes [Figure 2a] and [Figure 2b]. Sporadic inclusion body myositis (n=2): Both were male patients and were 65 and 70 years old respectively. Both presented with progressive proximal and distal muscle weakness, dysphagia and neck flexor weakness. The CPK was mildly elevated. Both the patients were on immunosuppressive therapy. Muscle histology showed rimmed vacuoles, lymphocytic infiltrate and hypertrophic fibers. The MHC class I antigen showed positivity along sarcolemma of single and small groups of non-necrotic muscle fibers. The intensity of staining was weak [Figure 3a] and [Figure 3b]. Dystrophies (n=20): The patient with DMD was an eight-year-old male and the patients with BMD included three male patients with ages ranging from 8-33 years (mean 23 years). The CPK was elevated in all patients and diagnosis was confirmed by immunostaining with dystrophin 1, 2, 3. Alpha sarcoglycanopathy included four male patients with age ranging from 6-11 years (mean 9.2 years). Gamma sarcoglycanopathy included one 24-year-old male patient. All of them had history of consanguineous parentage and elevated CPK levels. Diagnosis was confirmed by immunostaining with dystrophin 1, 2, 3 and sarcoglycans α, β, γ, δ and merosin. The patients with the diagnosis of Limb girdle dystrophies unclassified included seven male and two female patients with age ranging from 12-45 years (mean 24 years). All of them had history of consanguineous parentage and elevated CPK levels. Immunostaining was not contributory to diagnosis. The FSHD patient was a 20-year-old male with no family history and wasting of biceps, triceps and facial muscles. The CPK was mildly elevated. The myotonic dystrophy patient was a 48-year-old male with proximal and distal muscle weakness, dysphagia and myotonia. He had positive family history. All muscle biopsies of dystrophy showed inflammation on Hematoxylin and Eosin [Figure 4a]. The MHC class I antigen was negative in muscle fibers in all types of dystrophies and showed cytoplasmic positivity only in regenerating fibers. Muscle fibers adjacent to lymphocytic aggregates also were negative for MHC [Figure 4b]. There was no correlation of intensity of staining to inflammation or regenerative activity. Normal muscles (n=5). Muscle biopsies done for various other indications and histologically normal served as control group. There were two males and three females with ages ranging from 20-40 years (mean 25 years). The MHC class I antigen was negative in all muscle fibers and was positive in capillaries only. Discussion The diagnostic criteria for IIM proposed by Bohan and Peter do not distinguish polymyositis from inclusion body myositis and certain other dystrophies. [1],[2] Various other criteria proposed later were not validated. [3],[6],[7] Hence diagnostic criteria rely on histopathology and immunopathology. [3] This study used MHC immunostaining in DM, PM, IBM, various forms of dystrophies and controls and studied its utility as a diagnostic tool. Dystrophies were studied because the differential diagnosis of IIM is most often with dystrophies. Polymyositis and sIBM are muscle-specific antigen-mediated disorders. Cytotoxic T-cells target muscle fibers expressing MHC Class I antigens resulting in muscle necrosis. Expression of MHC Class I antigens on muscle fibers is an essential prerequisite for immunological interactions between muscle fiber and autoinvasive CD8 positive T-cell. [8] The MHC class I antigen expression on all muscle fibers is characteristic of PM while only the fibers that show partial invasion by CD8 lymphocytes in sIBM show the expression. [9] In the present study, both PM and sIBM showed positivity for MHC Class I antigen along sarcolemma in non-necrotic fibers surrounded by inflammatory cells and small groups of fibers. The fibers in sIBM showed less intense positivity as compared to PM. Dermatomyositis is an autoimmune humoral-mediated microangiopathy. The MHC Class I antigen expression was seen in the regenerating fibers, predominantly in the perifascicular area, in the present study. The MHC class I antihen expression in perifascicular muscle fibers and in areas of capillary loss may represent the triggering of MHC expression by a nonspecific cellular stress reaction, probably low-grade ischemia. [9] Interleukin-1 (IL-1) is mainly expressed by endothelial cells of capillaries in inflammatory myopathies and IL-1 helps to upregulate MHC Class I antigen. [10] The various forms of dystrophies included in this study showed uniform absence of MHC Class I antigen staining of muscle fibers including the fibers adjacent to inflammatory cell infiltrates. Regenerating fibers when present showed diffuse cytoplasmic staining. The dystrophies, especially FSHD, sarcoglycanopathies and other LGMD which have overlap features clinically, showed distinct features on MHC staining. There were no false positives in this study, though the number of patients in each group was small. In the large series reported by van der Pas et al. , on the diagnostic value of MHC Class I antigen staining in IIM, there were false positive results in 11% biopsies from muscular dystrophy and 4% of those with a miscellaneous neuromuscular disease. [3] Among the idiopathic inflammatory myopathies, polymyositis is an uncommon and frequently misdiagnosed disorder. [11] It is overdiagnosed as an entity among IIM for sIBM and DM without skin rash. It often poses diagnostic problems with dystrophies. Histopathology can be complemented with MHC staining in the diagnosis of IIM and in the differentiation from other muscle disorders. There were very few studies which used MHC staining in the diagnosis of IIM. [7],[9],[12],[13] van der Pas et al. , studied 208 muscle samples with MHC Class I antigen staining and found that there was no expression in congenital, metabolic, neurogenic disorders or healthy controls. Expression of MHC Class I antigen was found in 67% of muscle biopsies from patients with DM, in 61% with PM and in 96% with IBM. There were 15% false positive and 6.25% false negative results in their study. The authors emphasized the utility of staining muscle biopsy in patients on immunosuppressive therapy for less than four weeks and in absence of inflammatory infiltrates. The results of our study were similar to the study of van der Pas. [3] The recent study by Jain et al. , [5] also evaluated the use of MHC class I antigen in the diagnosis of IIM with a sensitivity of 88.7% and specificity of 89.1%. The MHC Class I antigen positivity was identified in 100% cases of DM and sIBM and 81.6% cases of PM. This is comparable to our study. The intensity of staining was variable in all cases, with seven cases of PM (particularly in the absence of inflammation) showing negativity. Moreover, two of the three cases of IBM showed strong positivity. This is in contrast to our study. Similar to our study, the majority of controls in their study showed lack of MHC class I antigen expression. The present study, though limited by numbers, holds promise that MHC Class I antigen expression can be used as a complementary diagnostic tool in the diagnosis and differential diagnosis of IIM. Acknowledgment Authors wish to acknowledge Mr. Madhavan for technical support.References
Copyright 2008 - Neurology India The following images related to this document are available:Photo images[ni08089f4.jpg] [ni08089f3.jpg] [ni08089t1.jpg] [ni08089f2.jpg] [ni08089f1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}