 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
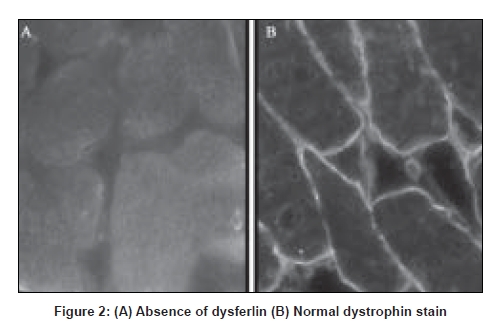
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 388-390 Case Report Twenty-two year follow-up of an Indian family with dysferlinopathy-clinical, immunocytochemical, western blotting and genetic features Khadilkar Satish V, Singh Rakesh K, Agarwal Pankaj, Krahn Martin, Levy Nicolas Department of Neurology, Grant Medical College and Sir J J Group of Hospitals, Byculla, Mumbai Correspondence Address:110, New Wing, First Floor, Bombay Hospital, 12 New Marine Lines, Mumbai-400 020 khadilkar@vsnl.com Date of Acceptance: 30-Sep-2008
Code Number: ni08094 Abstract Long-term observations over a period of 22 years in an Indian family with primary dysferlinopathy are recorded, defining phenotypic variability. In the propositus, the dystrophy began distally in the tibialis anterior muscles, before involving the gastrocnemius. Transient painful calf hypertrophy, followed by calf wasting was observed. The proximal lower and upper limbs weakened after three to four years. The younger sibling presented with the proximo-distal phenotype. Both patients showed very high creatine kinase values early into the illness. Disease progression was slow. The younger sibling lost ambulation 14 years after onset, while the elder one remains ambulatory 22 years into the illness. Muscle biopsy showed dystrophic features and absence of dysferlin. Monocyte western blotting confirmed absence of dysferlin. Genetic analysis detected a heterozygous mutation in Exon 54 [c.6124C>T] in the DYSF gene. This is the first family with a diagnosis of dysferlinopathy supported by genetic data, reported from India.Keywords: Dysferlinopathy, India, phenotypic, variability Introduction Dysferlinopathies encompass a spectrum of autosomal recessive neuromuscular disorders caused by mutations in the gene encoding dysferlin ( DYSF ; MIM# 603009, 2p13, GenBank NM_003494.2). [1],[2] Dysferlin is a large protein involved in membrane repair and vesicle trafficking and interacts probably with important immunological pathways. Dysferlin deficiency causes two main phenotypes: a distal Miyoshi myopathy (MM) and limb girdle muscular dystrophy Type 2B (LGMD2B). [1],[2] Both MM and LGMD2B show onset in young adults, slow course of the disease, and massive increase in serum creatine kinase (CK) level. However, MM affects the posterior compartment of the legs, while LGMD2B affects the pelvic girdle at onset. Although a few reports of Indian patients with dysferlinopathy exist, [3] no genetically analyzed case has yet been described from India. Long-term clinical follow-up is generally unavailable, especially in a case supported by genetic data. We describe an Indian family with immunocytochemically confirmed dysferlinopathy, further supported by DYSF mutational data, with observations on the clinical pattern of disease progression over 22 years. Case Report The family includes two affected brothers presently aged 43 and 40 years, of non-consanguineous parentage. They have been under observation for 22 years. The elder brother (initial index case) was initially seen at the age of 21 with complaints of weakness of foot dorsiflexion along with bilateral calf pains, of two years duration. Examination confirmed weakness of tibialis anterior muscles. Gastrocnemius muscle power was normal at this stage. Power in proximal lower limbs, trunk and upper limbs was normal. Reflexes and sensations were normal. CK was 4036 IU/L and electrophysiology showed myopathic changes. Serum TSH , vitamin B12, calcium was normal. Over the next two months, calf pains subsided. However, gradually calf wasting began to become apparent and foot drop increased. By 25 years of age, hip girdle, and later, shoulder girdle muscles started to become weak. A pattern of selective weakness became apparent with the tibialis anterior, gastrocnemius, hamstrings, hip adductors and iliopsoas being weaker than other muscles. At this stage, the biceps showed a ′lump′ [Figure 1]. In 1990, the patient′s younger brother, then aged 22, was initially examined. He had experienced difficulty rising from the ground for the previous year and a half. On examination, proximal lower limb weakness following a similar pattern as in the elder sibling was seen. The gastrocnemius and tibialis anterior muscles were mildly weakened. On enquiry, unlike his brother, he did not have any early calf pain. CK was elevated to 18344 IU/L and electrophysiology was myopathic. Electrocardiogram and 2D echocardiograms were normal in both patients. Muscle biopsy from hamstring muscles showed dystrophic features with completely absent dysferlin on immunocytochemical staining [Figure 2]. Monocyte western blotting showed absent dysferlin. Analysis of all 55 DYSF coding exons and flanking intronic boundaries was done in the index patient, using genomic DHPLC and subsequent direct sequencing of abnormally eluted fragments, as previously described. [4] Mutational analysis identified one heterozygous disease-causing mutation in Exon 54 [c.6124C>T;], predicted to cause the replacement of an arginine residue by a cysteine at position 2042 of the dysferlin protein. This mutation had been previously reported as pathogenic in other patients (data from the Leiden Muscular Dystrophy pages database; www.dmd.nl). Both brothers have been followed up regularly. Limb weakness has progressed very gradually in both. The differential weakness continues to be apparent with the tibialis anterior, gastrocnemius, hamstrings, hip adductors and iliopsoas remaining weaker than other muscle groups. While the elder brother is still independently ambulant at the present time and able to manage his daily job as a ward boy in the hospital with mechanical aids, the younger sibling has been confined to a wheelchair since 2003. Thus while the illness began at approximately the same age in both subjects, it has progressed more insidiously in the elder brother, while the younger sibling has shown relatively more rapid progression. Discussion Both subjects followed clinically for 22 years exhibit different phenotypic presentations of dysferlinopathy. The elder sibling presented with initial affection of the anterior tibial muscles. This mode of presentation of dysferlinopathy confirms to "distal myopathy with onset in the tibialis anterior" (DMAT) and though less common than the classic MM and LGMD2B phenotypes, is well described. [5] This initial involvement of distal muscles in the elder patient led us to consider a differential diagnosis of distal spinal muscular atrophy and other neurogenic processes, but the CK was much elevated, electrophysiology was myopathic and the true nature of the myogenic illness was further revealed as the full pattern of weakness unfolded over several years. The younger brother presented with simultaneous affection of the proximal and distal lower limb muscles. This increasingly recognized presentation has been referred to as the "proximo-distal" (PD) phenotype, and results either from MM after extension of disease to more proximal muscles, or from LGMD2B descending to the lower part of the lower limb, as was probably the case in our patient. In a large cohort of patients analyzed in France, the proximodistal form has been reported to occur in as high as 35% of dysferlinopathies. [6] Coexistence of two or three phenotypes (MM, LGMD2B, DMAT and others) within the same family or pedigree with dysferlinopathy is uncommon but known.[7],[8] Although no clear explanation exists, modifier genes or environmental factors have been thought responsible for such intrafamilial variation. In the present family, disease progression was also variable. The disease has progressed more insidiously in the elder brother, while the younger sibling has shown relatively more rapid progression, leading to wheelchair dependence 14 years from onset. An interesting feature observed in the elder subject was transient calf myalgia in the early phase of the disease. Later the calves wasted and pain subsided. This feature has been reported in patients with distal onset of dysferlinopathy [9] and is thought to be due to a transient inflammatory phase, similar to that described in FSHD . [10] CK levels in the early part of the illness were elevated to 4036 and 18344 IU/L respectively. In a patient presenting with distal motor symptoms, this massive increase of up to 50-100 times normal values is quite suggestive of MM and therefore of dysferlinopathy.[1] Over time, CK levels tend to decline and lose their informative value. Histopathological findings were in keeping with those described in the literature, consisting of fiber splitting, centralized nuclei and muscle fiber degeneration. In our patients, western blotting (WB) of peripheral monocytes showed absence of dysferlin. WB, a semi-quantitative technique, is aimed at assessing the amount of the protein of interest. In patients with dysferlinopathy, the protein can be quantitated from muscle or in an enriched pool of monocytes called CD14+. The test is convenient as it involves only blood sampling and the sensitivity is comparable to direct immunocytochemistry on muscle sections. [11] Routine mutational analysis of the DYSF gene is a challenging task, due to its large size and the large mutational spectrum. The DYSF coding sequence (6243bp) is divided into 55 coding exons and distributed on a genomic locus of more than 230 kb. To date, more than 300 different sequence variants have been detected along the coding sequence, including disease-causing mutations and non pathogenic polymorphisms (data from the Leiden Muscular Dystrophy pages database; www.dmd.nl). Noteworthy, currently used strategies for mutational analysis, either at the genomic or transcriptional level, allow for efficient detection of point mutations and small insertions and/or deletions within the coding sequence, which represent most of the DYSF disease-causing alleles. However, these strategies are insufficient for the systematic detection of some types of mutations, in particular "deep" intronic mutations which may affect normal splicing or regulatory regions, and exonic deletions and/or duplications. Such limitations partly allow explaining why both disease-causing mutations are found in only ~70 to 80% (depending on the screening procedure and inclusion criteria) of patients with dysferlin protein deficiency. In the index patient reported here, genomic mutational analysis identified only one of the two expected disease-causing mutations. However, in correlation with the clinical, histological and biochemical data, the identification of one DYSF disease-causing allele entirely supports the diagnosis of primary dysferlinopathy at the genetic level. In conclusion, this brief report documents a sib ship of Indian dysferlinopathy patients over a prolonged period of 22 years, highlighting the intrafamilial phenotypic variability. Acknowledgments The authors would like to acknowledge Miss Vιronique Labelle for expert technical assistance. References
Copyright 2008 - Neurology India The following images related to this document are available:Photo images[ni08094f2.jpg] [ni08094f1.jpg] |
| |||||||||

{kind=link}
{kind=link}