 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
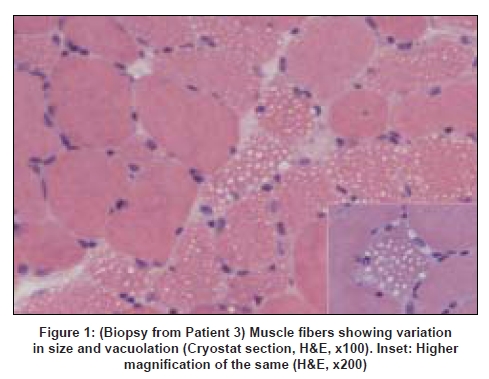
Neurology India, Vol. 56, No. 3, July-September, 2008, pp. 391-393 Case Report Lipid storage myopathies with unusual clinical manifestations Uppin Megha S, Sundaram C, Meena AK, Reddy Krishna Mohan, Reddy K Krishna, Vanniarajan A, Thangaraj K Nizam's Institute of Medical Sciences, Hyderabad Correspondence Address:Department of Pathology, Nizam's Institute of Medical Sciences, Punjagutta, Hyderabad - 500 082 challa_sundaram@yahoo.com Date of Acceptance: 13-Oct-2007
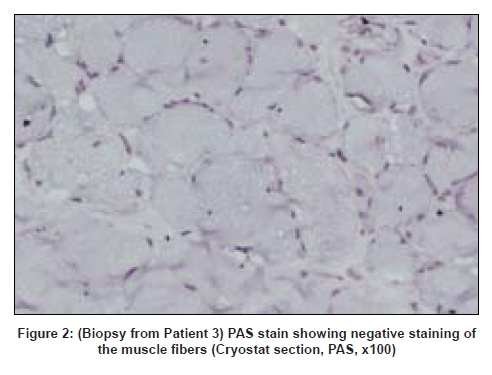
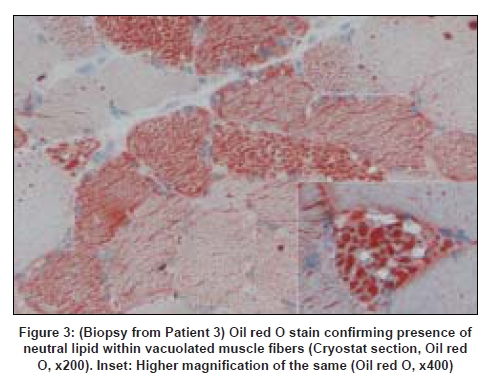
Code Number: ni08095 Abstract We describe the clinical presentation, course and pathologic findings found in three adult patients with lipid storage myopathy. Excessive lipid storage was found in Type 1 fibers of muscle. Clinical improvement on oral levo-carnitine therapy suggests the possibility of carnitine deficiency as the most likely etiology in two of the patients and one had mitochondrial myopathy confirmed on genetic analysis.Keywords: Carnitine therapy, lipid storage myopathy, muscle biopsy Introduction Lipid storage myopathies due to carnitine deficiency can present with varied clinical phenotypes that include chronic myopathy, cardiomyopathy, hepatic encephalopathy, organic aciduria and others. [1] Diagnosis is important because they are potentially treatable disorders. We report three such patients presenting with unusual clinical syndromes and successfully treated with oral carnitine therapy. Case Reports Patient 1: A 19-year-old female presented with acute abdomen and vomiting and was managed conservatively. She was readmitted after 20 days with similar complaints and diagnosed to have acute pancreatitis with elevated serum amylase and lipase levels. Blood glucose levels were mildly elevated (140 mg/dl). She complained of progressive proximal muscle weakness in the lower limbs after the first admission. The patient was re-admitted for the third time with severe breathlessness within 20 days of second discharge. The power in the lower limbs was markedly reduced (MRC Grade - 2/5 in proximal muscles of the lower limbs and 3/5 in distal muscles; 3/5 in both proximal and distal muscles of the upper limbs with significant neck muscle weakness). Deep tendon reflexes were not elicitable. A possibility of Gullian-Barre syndrome was considered. She was put on mechanical ventilator and was given intravenous immunoglobulin. Further investigations showed elevated creatine phosphokinase (CK) of 9000 IU/l with deranged liver function tests. Her respiratory symptoms improved, however, lower limb and neck muscle weakness showed only 50% improvement. A muscle biopsy was performed. Patient 2: A 32-year-old female presented with proximal muscle weakness in lower limbs followed by upper limbs over a period of 10 years. Power in lower limbs was markedly reduced (MRC Grade 2/5 in both proximal and distal muscles of lower limbs and 3/5 in upper limbs) and the deep tendon reflexes were not elicitable. Other system examination was within normal limits. The CK was 565IU/L and the electromyogram (EMG) showed myopathic pattern. Thyroid profile was within normal limits. Clinical possibility of limb girdle muscular dystrophy was suspected. Patient 3: A 47-year-old female presented with asymmetrical onset of proximal weakness starting in the right upper limb followed by both lower limbs and left upper limb over a period of 10 years with associated facial muscle weakness. On examination there was wasting of buccinator, bilateral supra and infraspinatus muscles with minimal wasting of small muscles of hand. Power according to MRC grading was 3/5 in proximal muscles of the upper limb on the right side and 4/5 on the left side. In the lower limbs the power was 3/5 proximally on both sides. Deep tendon reflexes were not elicitable. Examination of other systems was within normal limits. The CK was 1297 IU/L and EMG showed a myopathic pattern. Electrocardiogram, 2D echo, thyroid profile and lipid profiles were normal. A possibility of facioscapulohumeral dystrophy or limb girdle muscular dystrophy was considered. Muscle biopsy features: The biopsy was taken from the left vastus lateralis in all patients. In all three patients, biopsy showed fiber size variations and fine vacuolations, predominantly in Type I fibers [Figure 1] which were positive with oil red O [Figure 2] and negative with periodic acid Schiff stain [Figure 3]. Biopsy from Patients 1 and 3 showed regenerating fibers, indicating recovery from rhabdomyolysis. Biopsy from Patient 3 showed red ragged fibers on modified Gomori′s trichrome stain and increased oxidative enzyme activity. The light microscopic diagnosis in all three patients was lipid storage myopathy. As clinical diagnosis of mitochondrial disease was suspected in Patient 3, blood was submitted for genetic analysis. The entire mitochondrial genome was screened by polymerase chain reaction and sequencing methods. Two novel mutations in tRNA aspartic acid and Cytochrome B genes of mitochondrial genes were identified. Treatment and follow-up: Patients 1 and 2 were treated with Levocarnitine in the dose of 100 mg/Kg. Patient 1 is on follow-up for one and a half years and is performing her daily routine satisfactorily. The CK levels dropped down to 3500 IU/L. She discontinued the treatment on her own four months back and developed muscle weakness which improved on recontinuation of carnitine. The MRC grade on follow-up was 3/5 in proximal muscles of the lower limbs and 4/5 in distal muscles; 4/5 in both proximal and distal muscles of the upper limbs. Patient 2 is on follow-up for four months with definite improvement in her symptoms. The power improved to 3/5 in both proximal and distal muscles of lower limbs and 4/5 in upper limbs. The follow-up CK levels in Patient 1 dropped down to 3000IU/L and in Patients 2 and 3 are not available. Patient 3 was started on Levocarnitine 300 mg bd and there was no clinical improvement at the end of three months and she was subsequently started on Coenzyme Q 1 g/day two months back. Her response to treatment is awaited. Discussion Long chain fatty acids (LCFA) are the major energy substrate for muscle. Impairment at any level of mitochondrial beta oxidation pathway can cause lipid storage myopathy. The syndromes associated with lipid storage myopathies include carnitine deficiency (Type I lipid storage myopathy), carnitine-palmityltransferase (CPT) deficiency and pyruvate-decarboxylase deficiency. [2] Carnitine is an essential co-factor for the transport of LCFA into the mitochondria and modulates intracellular concentration of free coenzyme A in plasma and tissues. [3] Carnitine deficiency can be part of a number of inherited and acquired diseases. The clinical syndromes associated with carnitine deficiency include myopathic form with muscle weakness and systemic form with hepatic encephalopathy and muscle weakness, and progressive cardiomyopathy. [4],[5] Secondary carnitine deficiency may occur in patients with malnutrition, liver disease, chronic hemodialysis, mitochondrial disorders, organic acidurias, renal Fanconi syndrome, and valproate therapy. [6],[7],[8] The lipid storage myopathy in two of our patients was probably due to carnitine deficiency as they (1 and 2) responded to oral carnitine therapy and low-fat diet. Patient 1 probably had systemic form of carnitine deficiency with myopathy and hepatic encephalopathy precipitated by acute pancreatitis. Improvement, then relapse of muscle weakness after discontinuation of oral carnitine therapy and improvement after restarting the therapy confirms the diagnosis. Clinical syndrome of carnitine deficiency secondary to acute pancreatitis has not been reported earlier. Rhabdomyolysis and myoglobinuria with elevated CK and evidence of regenerating fibers on muscle biopsy were documented in Patient 1. Acute and recurrent myoglobinuria can occur in LCFA oxidation defects. [9] Myopathic form of carnitine deficiency as seen in Patient 2 is well documented in the literature. [6] It results when there is impairment of carnitine transport into the muscle. Though most of the reported cases in the literature are children, older patients as our patients are also reported in the literature. [10] The cause for the lipid storage in Patient 3 was probably secondary to mitochondrial disease. Red ragged fibers, increased oxidative enzyme activity and mitochondrial genetics support this. Mitochondrial disease causing lipid storage myopathy is described earlier. [11] When a metabolic myopathy involving fatty acid catabolism is suspected, muscle biopsy should be done only after doing serum carnitine and plasma free fatty acid. But in all our patients, muscle biopsy was the first clue for diagnosis. Diagnosis of lipid storage myopathy by muscle biopsy has been reported earlier from India. [12],[13] Due to lack of facilities, serum or tissue carnitine levels could not be done in our patients. In countries like India, where biochemical assay of carnitine is not readily available, muscle biopsy may provide an effective alternative diagnostic tool. The incidence of genetic defects of mitochondrial fatty acid beta oxidation is low. However, they represent an important cause of muscle disease. Characterization of mitochondrial beta oxidation disorders by genetic studies helps in diagnosis as in our patient. All patients with carnitine deficiency may not respond to therapy to the same extent. [14] Low-fat diet along with oral carnitine therapy was effective in two of our patients. Primary or secondary myopathic carnitine deficiency should be considered in the differential diagnosis of late onset myopathies as they are potentially treatable/ curable disorders. References
Copyright 2008 - Neurology India The following images related to this document are available:Photo images[ni08095f3.jpg] [ni08095f1.jpg] [ni08095f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}