 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 57, No. 2, March-April, 2009, pp. 119-125 Indian Perspective Neurology of acute organophosphate poisoning Gagandeep Singh, Dheeraj Khurana1 Departments of Neurology, Dayanand Medical College, Ludhiana, and 1 Postgraduate Institute of Medical Education and Research, Chandigarh, India Correspondence Address: Dr. Gagandeep Singh, Department of Neurology, Dayanand Medical College, Ludhiana - 141 001, Punjab, India. gagandeep_si@yahoo.co.uk Date of Acceptance: 08-Apr-2009
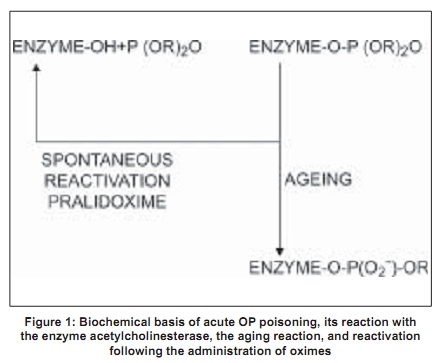
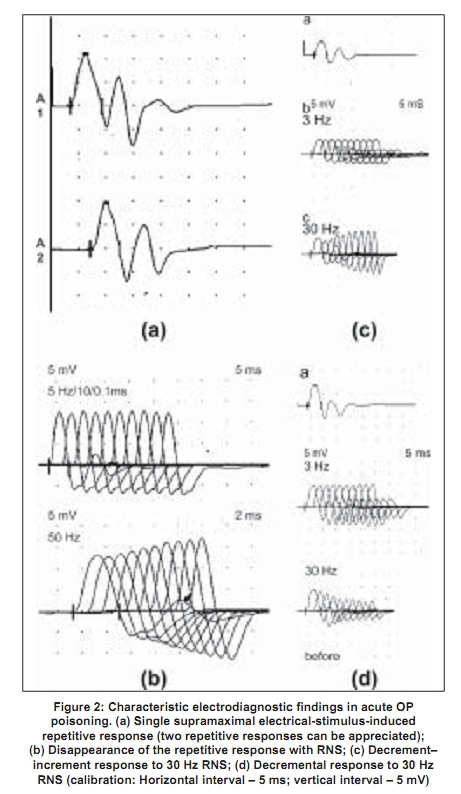
Code Number: ni09038 PMID: 19439839 DOI: 10.4103/0028-3886.51277 Abstract Acute organophosphate (OP) poisoning is one of the most common poisonings in emergency medicine and toxicological practice in some of the less-developed nations in South Asia. Traditionally, OP poisoning comes under the domain of emergency physicians, internists, intensivists, and toxicologists. However, some of the complications following OP poisoning are neurological and involve neurologists. The pathophysiological basis for the clinical manifestations of OP poisoning is inactivation of the enzyme, acetylcholinesterase at the peripheral nicotinic and muscarinic and central nervous system (CNS) nerve terminals and junctions. Nicotinic manifestations occur in severe cases and late in the course; these comprise of fasciculations and neuromuscular paralysis. There is a good correlation between the electrophysiological abnormalities and the severity of the clinical manifestations. Neurophysiological abnormalities characteristic of nicotinic junctions (mainly neuromuscular junction) dysfunction include: (1) single, supramaximal electrical-stimulus-induced repetitive response/s, (2) decrement-increment response to high frequency (30 Hz) repetitive nerve stimulation (RNS), and (3) decremental response to high frequency (30 Hz) RNS. Atropine ameliorates muscarinic manifestations. Therapeutic agents that can ameliorate nicotinic manifestations, mainly neuromuscular, are oximes. However, the evidence for this effect is inconclusive. This may be due to the fact that there are several factors that determine the therapeutic effect of oximes. These factors include: The OP compound responsible for poisoning, duration of poisoning, severity of poisoning, and route of exposure. There is also a need to study the effect of oximes on the neurophysiological abnormalities.Keywords: Atropine, neurophysiology, organophosphate poisoning, oximes, pralidoxime, repetitive nerve stimulation Introduction Organophosphate (OP) compounds are a large group of compounds having potential to irreversibly inhibit the cholinesterases, acetylcholinesterase, and neuropathy target esterase (NTE), in humans and animals. The OP compounds are not only used as insecticides and pesticides, but also used as chemical warfare agents, petroleum additives, and industrial plasticizers. Serious human exposure leads to both muscarinic (cholinergic) hyperstimulation and nicotinic receptor stimulation. Few OPs also inhibit NTE irreversibly. Historical Aspects Organophosphate compounds were known to exist since early 1800s, when Lassaigne synthesized OP compounds. However, the earliest recorded description of their synthesis was by de Clermont in 1854. Subsequently, Michaelis in Germany and Arbusov in Russia described synthesis of a large number of OP compounds in early 1900s. [1] However, the toxic effects of OP compounds were not recognized until 1932, when Lange and von Kruger described the toxic effects of OP vapors. In Germany, Schrader's deep involvement in the systematic research of OP compounds led to the development of more than 2000 OP compounds including sarin, parathion, and paroxon. Sadly however, given the prevailing scenario at the time, World War II, Schrader's work was mainly oriented toward the synthesis of chemical warfare agents. [2],[3] However, after the war, the pharmacological properties of these agents were published leading to the realization of their potential use as insecticides and pesticides. This eventually led to the development of many more new OP compounds and their main use as insecticides and pesticides worldwide. Contemporaneous to the development of OPs as insecticides, another group of OP compounds were developed for industrial use. These compounds are triaryl esters of phosphoric acid (e.g. tricresyl phosphate) and are now used as plasticizing agents, lead scavengers in gasoline, and as additives to hydraulic lubricants. Tri-orthocresyl phosphate was implicated in the enormous epidemic of Jinger-Jake paralysis in people who consumed contaminated alcoholic extract of Jamaican ginger in the United States in 1930s. [3] Epidemiology Poisoning with OP compounds is a worldwide phenomenon. An estimated three million cases of pesticide poisoning with 2000 deaths occur worldwide, each year. [4] According to the World Health Organization (WHO), one million serious unintentional poisonings occur every year and an additional two million people are hospitalized for suicide attempts with pesticides. [4] In India, OP compounds are among the most commonly used agents for suicidal poisoning. [5] Systematic community based data on the epidemiology of poisoning are not available from India. Hospital-based data suggest that barbiturates and copper sulfate were the commonly used agents in the years, 1972-1977; however, later they were replaced by OP compounds and aluminum phosphide. [6] In 1995, National Poison Information Center (NPIC) was established at the All India Institute of Medical Sciences, New Delhi. Data on the pattern of poisonings in North India accumulated at this center suggest that suicidal poisoning with house-hold agents is the most common modality of poisoning. [7],[8] The common house-hold agents included OPs, carbamates, pyrethrinoids, rodenticides, detergents, and corrosives. Agricultural pesticides accounted for 12.8% all cases of poisonings. Likewise, OPs caused most self-poisoning deaths in South and Central India. [9],[10],[11],[12] In a study from Andhra Pradesh, two-thirds of the patients were young adults aged less than 30 years, more than half were males, and attempted suicide was the most common intent for poisoning. Majority of deaths were due to poisoning with monocrotophos and endosulfan (an organochlorine). [11] Another study from Sri Lanka showed that young age, lower socioeconomic strata, unemployment, unstable emotional relationships, psychiatric disorders, and alcohol abuse were the risk factors associated with self-intentional or suicidal pesticide poisoning and suicidal intent accounted for almost 85% cases of pesticide poisoning. [13] Intoxication due to OP compounds may also occur by accidental exposure to agricultural pesticides while spraying, through skin and inhalational route. [14] Other potential modes of OPs poisoning include ingestion of adulterated fruit, flour, or cooking oil, and wearing contaminated clothing. [15] Choudary et al ., reported a food-borne outbreak of OP compound poisoning. [16] The kitchen, in which, food was prepared had been sprayed earlier with malathion. Among agricultural laborers, OP compound poisoning due to accidental exposure to agricultural pesticide spraying is not uncommon in India. Biochemical Basis Organophosphate compounds irreversibly inhibit the enzyme, acetylcholinesterase [Figure - 1]. [1] They combine with acetylcholinesterase to form a phopshorylated enzyme (enzyme-OP complex). This enzyme is functionally important at muscarinic and nicotinic nerve endings and also at central nervous system (CNS) synapses. Acetylcholinesterase inhibition results in prolonged action and excess of acetylcholine at the autonomic, neuromuscular, and the CNS synapses. The clinical manifestations of acute OP poisoning are the result of dysfunction in these synapses. Recovery from acute OP intoxication depends upon the reactivation of acetylcholinesterase-OP complex [Figure - 1]. Although, the reactivation of the enzyme occurs spontaneously, this is a very slow process. Rapid reactivation can be achieved by pharmacological agents such as oximes [Figure - 1]. The latter compounds are nucleophilic agents and act as acetylcholinesterase reactivators. However, the action of oximes is limited by the aging reaction, a time-dependent process that hydrolyses the enzyme-OP complex [Figure - 1]. As a result of aging, the enzyme is not susceptible to reactivation by oximes. As a general rule, the aging reaction occurs 48-72 hours after poisoning; hence at least theoretically, oximes are unlikely to provide therapeutic benefit beyond 48-72 hours after poisoning. In reality, different OP compounds have different aging half-lives. For instance, diethylphosphorylated acetylcholinesterase (a compound formed in poisoning by compounds such as parathion, chlorpyrifos, diazion, and chlorfenvinphos) has a long aging half-life and can be effectively reactivated by the oximes over sustained periods of time. [17],[18] On the other hand, dimethylphosphorylated acetylcholinesterase (a compound formed in poisoning by compounds like malathion, paroxanmethyl, dimethoate, and oxydemeton) has a very short aging half-life and is not susceptible to the reactivating action of oximes over longer periods of time. [17],[18],[19] Cholinergic transmission failure in OP poisoning involves both the muscarinic and nicotinic receptors and synapses. Acetylcholinesterase can be easily inhibited at the muscarinic synapses and nerve endings. However, failure of nicotinic transmission requires inhibition of at least 80% of the synaptic acetylcholinesterase. [1] This is the basis for the nicotinic syndrome (intermediate syndrome) being seen later in the course of OP poisoning (usually, following the acute muscarinic syndrome). Furthermore, the nicotinic syndrome occurs only in severe poisoning. [1],[20],[21],[22] Clinical Features The clinical features of acute OP compound poisoning can be categorized as follows: A muscurinic syndrome, a nicotinic syndrome, and a CNS syndrome. In addition, some of the patients may develop delayed peripheral neuropathy. Muscarinic syndrome The clinical features include rhinorrhea, bronchoconstriction, bronchorrhea, pulmonary edema, salivation, nausea, vomiting, abdominal cramps, defecation, sweating, bradycardia, urination, miosis, conjunctival hyperemia, blurred vision, and headaches. [21] The onset is immediately after exposure and the symptoms may last up to several days, depending upon the severity of intoxication. Nicotinic syndrome This syndrome usually follows muscarinic syndrome and precedes delayed neuropathy. For this reason it is referred to as 'intermediate syndrome'. Hyperstimulation of the neuromuscular junction by acetylcholine initially results in fasciculations, later followed by neuromuscular paralysis which may last for 2-18 days. [20],[22] The paralysis usually involves the ocular, bulbar, neck, proximal limb, and respiratory muscles in that order of severity. Central nervous system syndrome Central nervous system is uncommonly involved in acute OP poisoning and occurs with OP compounds that cross the blood-brain barrier. [22],[23] The manifestations include depressed mental status and central respiratory drive. In severe poisoning, patients may have convulsive seizures. Organophosphate-induced delayed neuropathy A delayed distal motor-sensory neuropathy may occur 2-4 weeks after poisoning with a very limited number of OP agents. A detailed discussion of the OP-induced delayed neuropathy is beyond the scope of the manuscript. Diagnosis Diagnosis of OP poisoning is based on the characteristic clinical features and history of exposure to a known OP compound. Estimation of serum or RBC cholinesterase level and electrodiagnostic tests are helpful in confirming the diagnosis. Clinical features of OP poisoning appear when RBC cholinesterase activity is < 75% of normal and in clinically overt poisoning it is usually < 10%. [1] In general, however, serial studies have failed to document a strict relationship between levels of serum cholinesterase and the severity of clinical manifestations and prognosis. [24] Clinical neurophysiology In nicotinic (intermediate) syndrome there is failure of transmission at the neuromuscular junction. Electrodiagnostic studies of neuromuscular transmission can be utilized to characterize and evaluate this disorder. [22],[24] In brief, the neurophysiological abnormalities include:
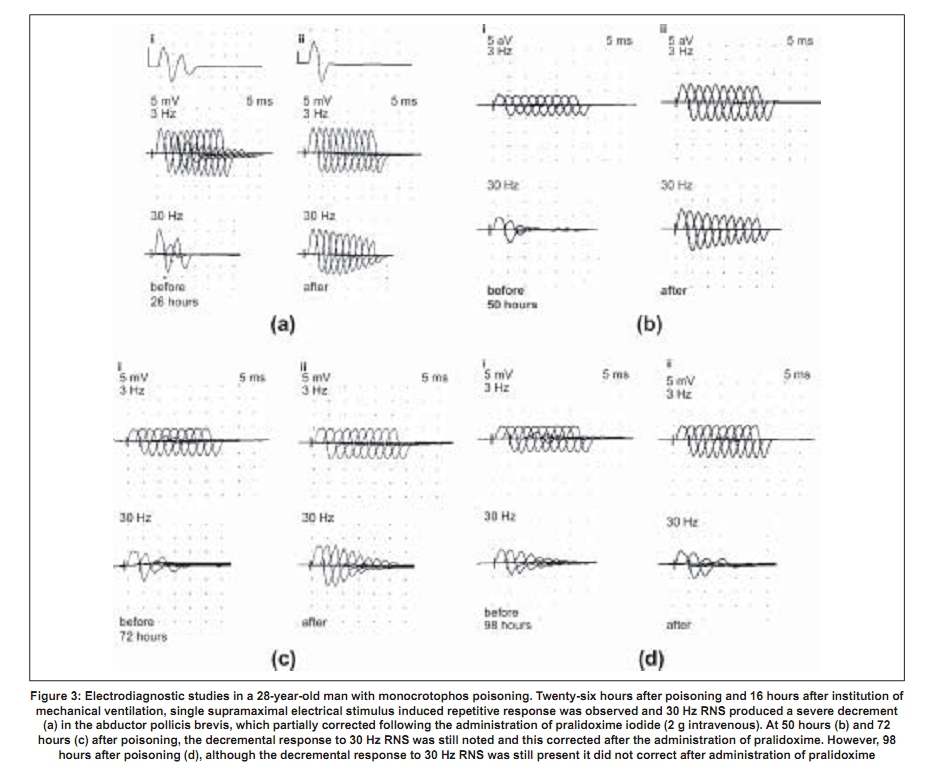
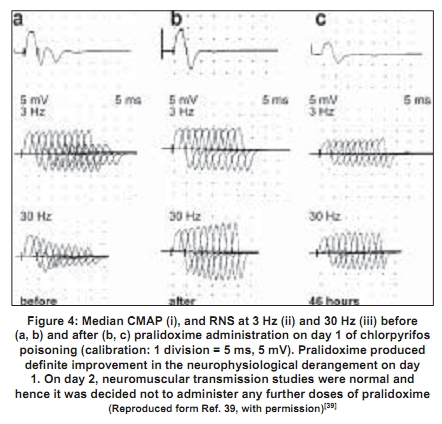
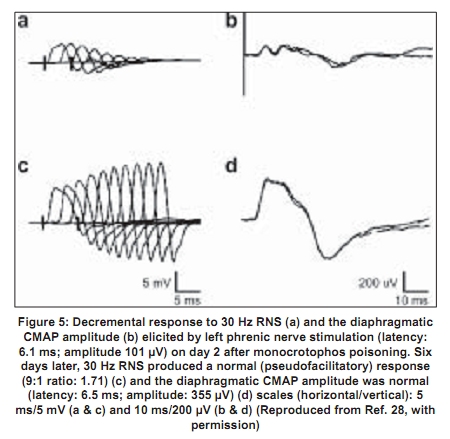
Repetitive response characteristically occurs in a resting muscle and disappears or decreases in amplitude after exercise and repetitive stimulation [Figure - 2b]. More than one repetitive response may be seen. Repetitive action of excess acetylcholine at the motor end plates is responsible for this electrodiagnostic finding. Decrement-increment response occurs at high rate (30-50Hz) RNS and is characterized by an initial dip in the amplitude of the compound muscle action potential and then a rise in the amplitude of the subsequent compound muscle action potentials (CMAPs). In vitro microelectrode studies suggest that the decrement-increment response represents a depolarization type of neuromuscular blockade. [25],[26] The decremental response is characterized by a progressive fall in the amplitude of the successive CMAPs elicited by a train of supramaximal electrical stimuli delivered to the motor nerve at 30-50 Hz. The ratio of the amplitudes of the ninth CMAP to the first CMAP (9:1 ratio) is arbitrarily taken as an objective marker of the severity of the decremental response. [1] It represents a desensitization type of neuromuscular blockade. [25],[26] It correlates with the presence of clinically recognizable intermediate syndrome. [24],[25],[26] It can be considered as an electrodiagnostic marker for the intermediate syndrome. [24] The electrodiagnostic abnormalities of acute OP poisoning are influenced by a variety of agents including pancuronium, oximes, and magnesium sulfate. Pancuronium paradoxically ameliorates the electrodiagnostic abnormalities. [27] The neurophysiological phenomenon is not associated with clinical improvement and can be explained by the blocking of acetylcholine receptors by pancuronium, a competitive neuromuscular blocking agent, thus preventing the OP-induced depolarization blockade. Organophosphate-induced respiratory failure - Clinical neurophysiology Respiratory failure is an important manifestation of the acute OP poisoning [20],[28],[29],[30] and is multifactorial: Respiratory muscle weakness in, particularly the diaphragm, central respiratory depression, and pulmonary factors (pulmonary edema, bronchoconstriction, bronchorrhea).[28] Electrodiagnostic tests can be of help in differentiating between neuromuscular and nonneuromuscular causes of respiratory failure in acute OP poisoning. Low amplitude of the diaphragmatic CMAP correlates well with the need for assisted mechanical ventilation and the reduced amplitude improves with amelioration of the neuromuscular transmission deficit [Figure - 3a,b,c,d]. [28] Treatment The initial management of acute OP poisoning includes cardiorespiratory stabilization, removal of clothes (a possible source of continued exposure in occupational intoxication), irrigation of skin and eyes, and gastric lavage to minimize absorption of the OP compound. The mainstay of treatment in acute OPC poisoning is atropine, a muscarinic acetylcholine receptor antagonist. Atropine antagonizes muscarinic symptoms and signs but has little or no effect on nicotinic features. Nicotinic features can probably be treated by oximes (including pralidoxime and obidoxime). Of the four different preparations of pralidoxime, pralidoxime iodide and pralidoxime chloride are available in India. The other two preparations are pralidoxime mesylate and pralidoxime metisulfate. There has been no head-to-head comparison of any of the four agents. Role of cholinesterase reactivation Oximes have been shown to have protective effect in experimental nerve gas poisoning. [30] The use of oximes in human acute OP poisoning has been largely empirical.[31],[32] A recent study did not find any advantage of continuous infusion of 12 g/day of pralidoxime for three days over single-dose paralidoxime. [33] The same group of workers later compared the effect of conventional dose of pralidoxime to placebo. [34] Pralidoxime administration was found to be associated with higher incidence of intermediate syndrome, ventilatory requirement, and mortality. However, methodological issues in these two trials may have influenced the interpretation of the negative results represented in these trials. The dosage employed were below the WHO-recommended dose. [35] Studies of oximes in experimental animals (sarin models) have shown to improve neuromuscular transmission. The dosage of pralidoxime found to be effective in these models was 4 mg/L. [23] Extrapolating these results to human beings, the WHO-recommended dose of pralidoxime is 30mg/kg bolus followed by 8mg/kg/hour infusion. [19] The other limiting aspect of these studies were: (1) the randomization and masking techniques were not adequately described in both the studies [33],[34] and (2) in the latter study the intervention arm included patients with more severe poisoning which could explain the increased frequency of intermediate syndrome, ventilator requirement, and mortality in the intervention arm. [36] Discrepancies between the robust rationale for the use of oximes and the failure to demonstrate their efficacy in clinical trials can be understood if the limitations of oxime action are appreciated. One of the major limitations of oximes is their short duration of action. The half-life of pralidoxime in human OP poisoning varies between 1.5 and 3.5 hours. [23],[37] Thus, a single dose of pralidoxime is unlikely to improve the clinical outcome in severe OP poisoning. Another major limitation is that conventional doses of oximes are ineffective in extremely severe OP poisoning. Willems et al ., studied the relationship between OP compound concentration and oximes effectiveness and found that pralidoime may not reactivate ethyl and methyl parathion levels greater than 30 µg/L. [37] Organophosphate compounds also differ in their susceptibility to reactivation by different oximes. For instance, obidoxime is superior to pralidoxime in dimethylphosphoryl poisoning. [17],[18] A major reservation about oxime administration in acute OP poisoning derives from the knowledge of the aging reaction, a time-dependent biochemical process that involves the formation of a negatively charged monophosphoric acid residue-enzyme complex. The aged complex is resistant to reactivation on account of its chemical structure [Figure - 1]. [19] Given the several limitations of the use of oximes in acute OP poisoning, there is a need for the development of a dynamic parameter that can be monitored, in order to guide decisions such as continuation of oxime therapy and its dosage. Neuromuscular transmission studies are an objective parameter that can be used to follow up patients with acute OP poisoning. Neurophysiological studies correlate well with clinical findings in acute OP poisoning. In particular, the decremental response to 30Hz RNS is a marker for the 'intermediate syndrome'. [24] The amelioration of the 30Hz decremental response is therefore indicative of an improvement in the neuromuscular transmission, both in pathophysiological and clinical terms. The authors' studies have shown that administration of oximes is associated with the disappearance of the decremental response [Figure - 4] and [Figure - 5]. The decremental response reappears later as the effects of the oxime wear off. The authors routinely use and recommend neuromuscular transmission studies to monitor the beneficial effects of oximes and to guide decisions such as the duration and dose of pralidoxime in individual cases. [27] It is unlikely that any fixed-dose regimen of pralidoxime can be recommended. The dose and frequency and duration of administration should be individualized and may be influenced by factors such as the individual OP agent involved, the quantity of exposure and duration of poisoning, and existing cholinesterase levels, and most significantly the existing neurophysiological findings. 'Tailored pralidoxime administration under electrodiagnostic data guidance' would be preferable rather than administration of a fixed-dose regimen. There are few reports with the use of other agents like bicarbonate and magnesium sulfate. Magnesium sulfate acts at the neuromuscular junction by reducing concentration of acetylcholine. Two studies have shown reduction in mortality with magnesium sulfate and favorable changes in electrophysiological parameters.[27],[38] A randomized study at one of the authors' center comparing magnesium sulfate with placebo in acute OP intoxication did not show any differences in the outcome with addition of magnesium sulfate to standard-treatment regimen. Eighteen patients with OP poisoning presenting within 24 hours of the intoxication were administered magnesium sulfate (4 gm q 6 hourly for 48 hours, intravenous) or placebo, in addition to atropine and pralidoxime. No significant difference in the clinical and electrophysiological outcomes was observed (Dr. D. Khurana, personal communication).[39] References
Copyright 2009 - Neurology India The following images related to this document are available:Photo images[ni09038f2.jpg] [ni09038f1.jpg] [ni09038f4.jpg] [ni09038f3.jpg] [ni09038f5.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}