 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 57, No. 2, March-April, 2009, pp. 126-133 Review Article Transverse myelitis spectrum disorders Lekha Pandit Department of Neurology, KS Hegde Medical Academy, Mangalore, India Correspondence Address: Dr. Lekha Pandit, Department of Neurology, KS Hegde Medical Academy, Deralakatte, Mangalore-575 018, Karnataka, India. panditmng@gmail.com Date of Acceptance: 05-May-2009
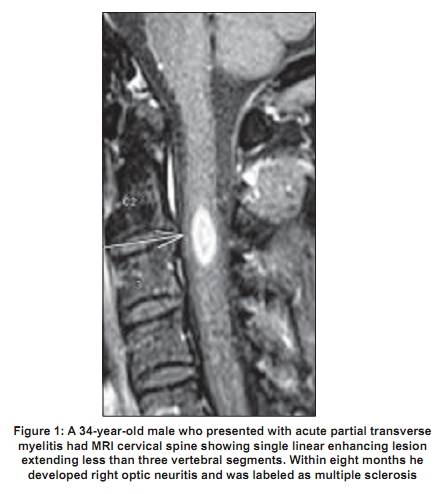
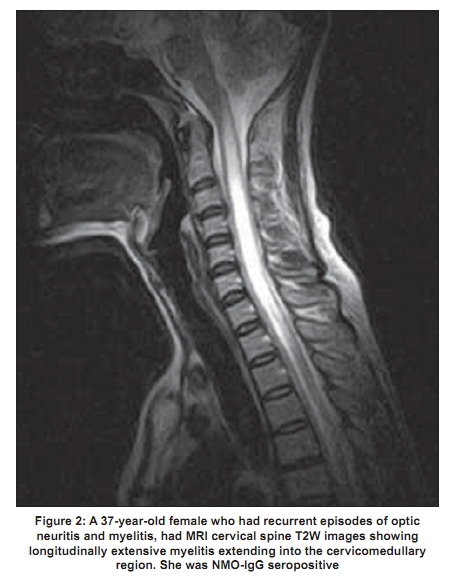
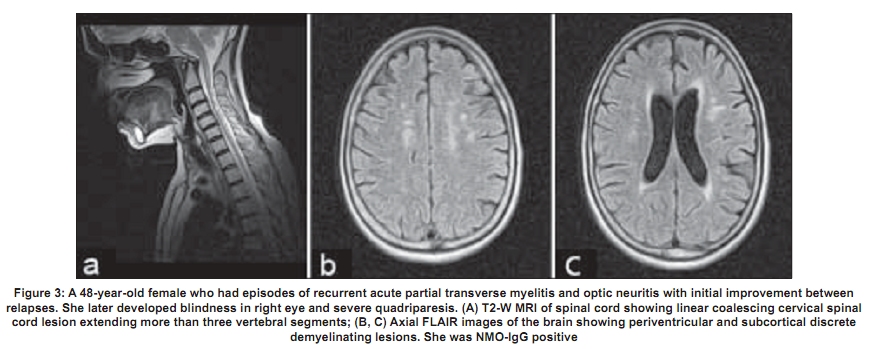
Code Number: ni09039 PMID: 19439840 DOI: 10.4103/0028-3886.51278 Abstract Acute transverse myelitis (ATM) is an inflammatory demyelinating disorder that affects the spinal cord focally resulting in motor sensory and autonomic dysfunction. Establishing the diagnosis of ATM is not as difficult as determining the possible etiology. There is a difference in the perception of ATM seen in the West as compared to developing countries. In the West multiple sclerosis (MS) is the most common inflammatory disorder of the central nervous system. An attack of ATM may be the beginning of MS. However, this may not be the case in developing countries where MS is uncommon. Most often transverse myelitis is monophasic and at best represents a site-restricted form of acute disseminated encephalomyelitis (ADEM). Traditionally the combination of optic neuritis and ATM, occurring as a monophasic illness would have been called as neuromyelitis optica (NMO). Changing concepts in the definition of NMO and the discovery of a biomarker, neuromyelitis optica immunoglobulin (NMO_IgG), has changed the way relapsing autoimmune disorders are being perceived currently. A variety of idiopathic inflammatory disorders such as Japanese form of optic spinal MS, recurrent myelitis, and recurrent optic neuritis have been brought under the umbrella of neuromyelitis spectrum disorders because of the association with NMO-IgG. Complete transverse myelitis accompanied by longitudinally extensive transverse myelitis which is seronegative for this biomarker has also been reported from several countries including Japan, Australia, and India. Thus, ATM is a heterogeneous disorder with a varied clinical spectrum, etiology, and outcome.Keywords: Neuromyelitis optica, NMO spectrum disorders, NMO-IgG, transverse myeltis Introduction Acute transverse myelitis (ATM) is a pathogenetically heterogeneous inflammatory disorder of the spinal cord. An incidence of 1-4 per 100,000 has been reported in Western literature. [1] Though no epidemiologic data are available from India, ATM has been reported as the major cause of noncompressive myelopathy. [2],[3] A variety of disorders can cause ATM and includes infections, para and postinfections, and vascular, neoplastic, paraneoplastic, collagen vascular, and iatrogenic irregularities. [4] Until recently, it was believed that all relapsing idiopathic inflammatory diseases were multiple sclerosis (MS). [4] With revisions in criteria for neuromyelitis optica (NMO), [5],[6] the traditional concept of NMO being a monophasic disorder has been set aside. It is now accepted that it is characterized by severe attacks of optic neuritis (OPN) and myelitis which spares the brain, especially early in its course. Unlike MS there is a specific biomarker for NMO, neuromyelitis optica immunoglobulin G (NMO-IgG). This is an autoantibody present in the serum of patients with NMO which distinguishes neuromyelitis from other demyelinating disorders. [7] NMO-IgG binds to aquaporin-4 which regulates water homeostasis in the central nervous system (CNS). [8] It has also been detected in some patients with recurrent myelitis associated with longitudinally extensive spinal cord lesions (LESCLs), optic spinal MS (OSMS) seen in Japan (Asian form of multiple sclerosis), recurrent isolated OPN, and OPN or myelitis associated with certain autoimmune disorders. [9] The transverse myelitis (TM) consortium [10] attempted to formulate a definition which would help to establish uniform diagnostic criteria and at the same time exclude other conditions that mimic this disorder. However, this definition does not help the clinician in deciding whether a given case has chances of converting to MS, whether it is a restricted from of acute disseminated encephalomyelitis (ADEM), or whether it belongs to the NMO spectrum of disorders. A clue to clinical diagnosis lies in the presentation: acute partial TM (APTM) or acute complete TM (ACTM). It also lies in its clinical course: monophasic or relapsing, and also in the clinical accompaniments such as OPN. The nature and extent of lesions on the magnetic resonance imaging (MRI) of the spinal cord and the presence and distribution of brain lesions are important. Lastly, the seropositivity for aquaporin-4 antibody, otherwise called NMO-IgG antibody, is important, though in the current scenario it is not feasible to do it routinely especially in developing countries. Definitions It is important that the nosology of terms used in relation to TM is agreed upon and commonly used. Lack of uniformity creates diagnostic confusions and overlap between disease entities. The classical example would be the term OSMS . This term was loosely coined to describe MS with clinical attacks confined to the spinal cord and optic nerve in the earlier literature from both Japan and India. With the advent of MRI, Japanese authors have used this term to describe recurrent severe TM and OPN which are accompanied by longitudinal spinal cord lesions on MRI. [11] This definition closely resembles the revised criteria for NMO. [4],[5] In addition, the term spinal MS (SMS) has also been used to describe recurrent TM. [12] Acute complete transverse myelitis Acute complete transverse myelitis [13] may be defined as an idiopathic inflammatory disorder causing symmetric spinal cord dysfunction below a specific level of cord function. The ensuing disability may be moderate to severe. Acute monophasic TM has been described in the background of variety of infections and vaccinations. It may also be seen in the setting of ADEM, in nearly a quarter of patients. [14] Severe forms of ATM have been described with other autoimmune disorders such as systemic lupus erythemtosis (SLE), Sjφgren's syndrome (SS), primary antiphospholipid antibody syndrome, sarcoidosis, and various forms of vasculitis. [9] It may be the initial manifestation in as much as 23% of patients later diagnosed to have SLE. [15] When ATM presents symmetrically with moderate to severe spinal cord dysfunction, the obvious implication is that chances of conversion to MS is remote. The conversion rate is less than 2% at five-year follow-up. [16] Rarity of ACTM in MS has also been highlighted in other studies. [17] Pediatric ATM is most often post infectious and may have a better outcome than adult patients. In addition, recurrence in myelitis and conversion to MS are rare. [18] Prognosis may be better for ATM occurring with ADEM than in isolated ATM. [19] Acute partial transverse myelitis In contrast to ACTM, APTM [20] may be defined as an asymmetrical or mild loss of spinal cord function. These patients may have patchy sensory impairment, mild to moderate weakness of asymmetric distribution, and occasional bladder dysfunction. Patients with APTM have greater chances of converting to MS. In one of the earlier studies on APTM, Ford et al , [21] have clearly shown that majority of patients presenting with asymmetric and patchy spinal cord dysfunction had converted to clinically definite MS (CDMS) within three years. Thirteen out of 15 patients (87%) who converted had abnormal brain MRI at onset of disease. When the initial brain MRI was normal, over a five-year follow-up period, approximately 20-30% converted to definite MS. [21] In patients presenting with APTM, detection of oligoclonal bands at onset or abnormal evoked potential studies were however not reliable predictors for conversion to CDMS. [22] Patients in this group are also not at high risk for developing NMO, [23] though some patients can experience mild relapse in spinal cord symptoms during the course of the illness. In conventional MS, also referred to as Western form of MS, spinal cord lesions are usually fewer than two vertebral segments and occupy less than one-half of a spinal cross-section, preferentially involving the peripheral white matter [Figure - 1]. [24] Neuromyelitis optica Neuromyelitis optica diagnostic criteria have been revised twice in recent times. [5],[6] NMO may be monophasic or recurrent. The most recent criteria require acute OPN and myelitis as absolute requirements. Alongside, two of the following supportive criteria are required: contiguous spinal cord MRI lesion extending over three vertebral segments, brain MRI not meeting diagnostic criteria for MS and NMO-IgG seropositive status. Evolution of criteria for diagnosis of neuromyelitis optica The standard definition of NMO (Devic's disease) was one of a monophasic disorder involving spinal cord and both the optic nerves, without involvement of the rest of the neuraxis. Though it was accepted that the underlying pathology was inflammatory, opinion was divided as to whether it represented a distinct disease, a variant of MS, or a postinfective restricted form of ADEM. [25],[26] In 1999 Wingerchuck et al , [5] put forward the first of their revised criteria for NMO. Three absolute requirements were OPN, acute myelitis, and no symptoms implicating other CNS regions. Fulfillment of at least one of three major supportive criteria was required: 1) normal brain MRI at disease onset or MRI not fulfilling MS imaging criteria; 2) spinal cord MRI showing a lesion extending over> 3 vertebral segments; and 3) cerebrospinal fluid (CSF) revealing> 50 WBC/mm 3 or> 5 neutrophils/mm. [3] Alternatively, fulfilling two of three minor supportive criteria (bilateral OPN, severe residual visual loss, or severe fixed postattack weakness) would suffice. In 2006, the NMO diagnostic criteria [5] were revised to include patients who had features of NMO but had additional sites of neurological dysfunction or brain MRI lesions not consistent with MS [Table - 1]. The most notable features of this revised criteria were obviously the inclusion of newly detected biomarker - NMO-IgG. Nonspecific brain lesions can develop in 60% of patients which immunohistochemically resemble spinal cord lesions of NMO. [27] MRI lesions typical of MS can develop in 10% of patients who otherwise fulfill criteria for NMO. In another 10%, white matter lesions can develop in aquaporin-rich periependymal regions such as hypothalamus and the periaqueductal brainstem. [28] Earlier it was thought that brain lesions in NMO were asymptomatic, but now it is accepted that some lesions may be symptomatic - for example, the nonautoimmune endocrinopathies reported in association with NMO. More than 90% of patients fulfilling the criteria have a relapsing course rather than monophasic and have recurrent OPN and myelitis. [29] Longitudinally extensive transverse myelitis Spinal cord lesion length has been emphasized as an important distinguishing factor between NMO and MS. Longitudinally extensive transverse myelitis (LETM) or LESCLs or long cord lesions (LCL) refers to idiopathic spinal cord inflammatory lesions extending more than three vertebral segments on the MRI. NMO-IgG seropositivity appears to be much more likely in patients presenting with lesions extending over multiple spinal levels compared with patients with small single-level cord lesions. [30] In conventional MS reported from the West, 3-12.5% of patients have been reported to have LETM. [24],[31] Longitudinal cord lesions are seen in 25% of Japanese patients with conventional MS. [32] In the Japanese form of OSMS, the incidence is much higher, 59%. However, it is worth noting that the Japanese definition of OSMS closely resembles that of NMO as mentioned earlier. Clinical features - Neuromyelitis optica versus multiple sclerosis Relapsing NMO has a 5:1 preponderance in women, though the monophasic variant is equally distributed among sexes. The mean onset of symptoms is later in NMO, most often in the third decade rather than the second (in MS). The severity of visual loss and the incomplete recovery is a hallmark of NMO. Patients are conscious of their visual loss. In MS, very often pallor of the optic disc detected on clinical examination or a delayed visual evoked response draws attention to the underlying OPN. Complete transverse myelitis and the incomplete recovery are other clinical features that should alert the physician. The occurrence of hiccoughs or respiratory failure during the course of myelitis should raise suspicion for NMO. [29] The course of the disease is distinctly different from MS. Relapse occurs within one year in 60% and within three years in 90%. [5] Within five years of disease onset, more than 50% of patients with relapsing remitting NMO are blind in one or both eyes or require ambulatory support (NMO spectrum disorders). The calculated five-year survival rate for NMO is 68%, all deaths are related to neurogenic respiratory failure. [33] In comparison the course in relapsing remitting MS is different. Attacks are mild with good recovery and permanent disability generally builds up over the years during the secondary progressive phase of the disease. A secondary progressive phase is rare in NMO. [34] The immunopathology of neuromyelitis optica and the neuromyelitis optica-IgG Neuromyelitis optica lesions are characterized by necrotizing lesions involving both gray and white matter of the spinal cord extending across many segments and resulting in cavitations. [35] Inflammatory infiltrates of active lesions in NMO have eosinophils and neutrophils, a feature unique to NMO and not seen in MS. Penetrating spinal vessels are thickened and hyalinized. [36] Immunoglobulin and compliment components are deposited in a characteristic vasculocentric rim and rosette pattern in active lesions. This pattern corresponds to normal expression of aquaporin-4 in the end feet of astrocytes. [37],[38] In contrast to MS lesions, in which aquaporin-4 immunoreactivity is increased, aquaporin-4 is absent in NMO lesions. [37],[39] The NMO-IgG, an autoantibody reported by Lennon and colleagues, was found to be 73% sensitive and 91% specific for 124 clinically defined NMO patients studied prospectively. [7] NMO-IgG binds to aquaporin-4 which is the main channel that regulates water homeostasis in the CNS. Subsequently, data from other European countries confirmed the sensitivity and specificity of NMO-IgG in differentiating NMO from MS. The authors have however admitted that 10-25% of patients with clinically diagnosed NMO were seronegative for NMO-IgG. NMO-IgG may also have a predictive value. Weinshenker et al , [40] evaluated 29 patients with a first attack of TM with an MRI lesion spanning three or more vertebral segments. Of 23 patients followed for one year, 9 were seropositive for the NMO-IgG autoantibody. Within one year, five of nine patients had a second event, involving recurrent TM in four patients and OPN in one patient. After 1-7 years of follow up, none of the 14 patients who were seronegative for the NMO-IgG autoantibody had a relapse of myelitis or OPN. Expanding spectrum of neuromyelitis optica A variety of allied disorders is grouped under the spectrum of NMO, based on the detection of NMO-IgG in the serum of affected patients reported from select centers. This includes Asian OSMS (reported from Japan), recurrent myelitis associated with LESCLs, recurrent isolated OPN, and OPN or myelitis in the context of certain organ-specific and nonorgan-specific autoimmune disorders such as SLE and SS. [9] Optic spinal multiple sclerosis The detection of NMO-IgG in select group of patients with OSMS from Japan, led to the conclusion that Japanese patients with OSMS may actually have NMO. In 12 out of 19 patients (65%) with OSMS and in 2 out of 13 patients with CMS, NMO-IgG was positive. [41] Features of Japanese OSMS include: (1) older age at onset, (2) female preponderance, (3) frequent relapses, (4) greater disability due to severe optic nerve and spinal cord damage, (5) fewer brain lesions detected by MRI, (6) LESCLs extending over many vertebral segments on spinal cord MRI, (7) marked pleocytosis and neutrophilia in CSF, and (8) absence of oligoclonal bands in CSF. [11] These observations are almost identical to the criteria proposed for NMO and are at variance with definitions used in other studies including from India and may explain the high degree of seropositivity in this Japanese series. Out of the original cases studied by Nakashima et al , [41] two of the cases labeled as CMS and seropositive for NMO-IgG on review were found to have LETM on MRI. This exemplifies the need for uniform diagnostic criteria and the fact that perhaps length of spinal cord lesions correlated best with seropositivity in Japanese OSMS. In a more recent study, Matsuoka and colleagues [42] have studied consecutive sera from 113 patients with MS which included OSMS (48) and CMS (54). Interestingly only 15 (27.1%) of OSMS showed seropositivity. When the MRI data were incorporated, it was found that 9 out of 48 OSMS patients had LETM and 5 of them were seropositive for NMO-IgG. Compared to the earlier Japanese study, NMO-IgG seropositivity was much lower. Kira et al , [7] commented on the obvious differences in seropositivity among Japanese patients. The series of Takahashi et al , [43] (20/22, 91% in NMO patients), Tanaka et al , [44] (60% in OSMS patients with LESCLs), and Matsuoka et al , [45] (11/31, 35% in OSMS patients with LESCL) had varying results for the NMO-IgG assay. On analysis of these studies, [7] it was found that there was no homogeneity in patient selection, those having LETM were lumped with patients having shorter cord lesions. In addition, some of the studies included only female [43] or predominantly female patients and were selected from an existing database rather than consecutive cases. Matsuoka et al , [42] who had the lowest seropositivity, studied consecutive patients and included both sexes. At one study center in Japan [7] the sensitivity of NMO-IgG detection assay was increased to 100% without any improvement noted in the detection rates. This eliminated the variation in seropositivity due to poor assay sensitivity. Recurrent myelitis Relapsing myelitis has been recognized as a condition distinct from MS. [45],[46] Recurrent CTM associated with longitudinal spinal cord lesions was more likely to be NMO-IgG seropositive in Western case series. [9] As mentioned earlier, NMO-IgG seropositivty after the first attack of ACTM predicts a relapsing course in more than 50% of patients within three years of the first attack. Myelitis associated with organ-specific and nonorgan-specific autoimmune disease Weinshenker et al , [47] observed NMO-IgG seropositivty in some patients with SLE and SS who had NMO or NMO spectrum disorders. In contrast, SLE and SS associated with systemic autoimmune disease and uncomplicated by NMO were seronegative for the biomarker. Pittock et al , [48] from the same group found a higher frequency of nonorgan-specific autoantibodies in NMO-IgG-positive patients than in NMO-IgG-negative patients. Nonorgan-specific autoimmunity was however seen equally in NMO seropositive and negative cases reported from a Japanese study of OSMS. [41] Neuromyelitis optica and neuromyelitis optica-IgG status in Indian patients Dastur and Singhal [49] described the autopsy findings of a patient who probably was the first pathologically proven NMO case from India. The patient, a woman who had clinical attacks of recurrent OPN and myelitis, died of respiratory failure and had autopsy finding confined only to spinal cord and optic nerves. NMO is obviously present in Indian population but the speculation is, how common is it? Hospital-based case series reported from India in the preMRI era [50],[51] are often quoted in Western literature on NMO, to support the notion that NMO is common in India. The basis for this speculation could be possibly the disproportionate involvement of spinal cord and optic nerve reported in Indian MS. Optic spinal MS, loosely described in most Indian literature as MS, with attacks confined clinically to spinal cord and optic nerve were seen in 20-60% of cases. [52] There is also paucity of oligoclonal bands in Indian MS. Jacob et al , [53] who reviewed published literature from India on NMO, identified 59 cases (9-24%), from hospital series reported from different parts of India. The authors opined that with more relaxed criteria for NMO, the frequency was likely to be higher. Bansal et al , in a case control study compared matched American and Indian patients and did not find an excess of NMO in their Indian patients. [54] The frequency of NMO in this MRI-supported study, in Indians was 6%. In a prospective study, using revised criteria for both MS and NMO and backed by MRI data, Pandit et al , [55] found only 5% of NMO in their patients while 43% of cases were OSMS. Recently, Pandit and colleagues analyzed NMO-IgG status in 78 consecutive cases obtained from their demyelination registry. [56],[57] In this study 63/78 (81%) patients belonged to the NMO spectrum. Longitudinally extensive transverse myelitis was seen in 51% and included all cases of NMO, ATM, and recurrent ATM. Neuromyelitis optica-IgG was positive in three female patients (3.8%), one each of NMO [Figure - 2], OSMS [Figure - 3], and recurrent ATM. Seropositive patients were all women and had late onset disease. Both patients with OPN had severe visual impairment and all three were wheel chair bound within five years of disease onset. Seronegative patients (36) had M/F ratio: 2:1 and had mild visual impairment. Assistance for mobility was required by 30% (12). The remaining patients recovered well between attacks and remained ambulant 4-7 years after disease onset. An earlier clinical study from India by Pradhan et al , [58] showed a similar outcome. They reported six patients, three male and female patients, each who had recurrent CTM and OPN. Brain MRI was normal, but in all, spinal cord MRI showed LETM extending 6-9 segments. These patients on follow up, 2-10 years later, were independently ambulant except for one patient who required a cane. It is clear that seronegative cases far exceed NMO-IgG-positive patients, in India. Additionally, the clinical behavior of patients in both studies quoted above distinguished them as separate entities. They had milder spinal cord and visual disturbances in spite of LETM, during a lengthy follow-up period. There are only few studies reporting low seropositivity [59],[60] among Caucasian patient populations. Given that the proponents of the concept of NMO spectrum disorders [9] admitted that 25% of patients tested by them were negative for NMO-IgG. There have been no studies analyzing these seronegative patients especially from the West. From Japan, Matsuoko and colleagues [42] in their landmark paper highlighted that a significant number of their OSMS patients were seronegative (65%). Optic neuritis in the seronegative cases was less severe. They had LETM which differed from seropositive patients. MRI lesions in the seronegative group appeared throughout cervical to thoracic cord as compared to seronegative patients who had lesions in the upper or midthoracic cord. On axial sections, seronegative patients had a holocord appearance compared to central gray matter involvement in seropositive patients. Other Japanese studies have also noted a lesser frequency of relapses and milder visual disturbances in their seronegative patients. [41],[44] These studies sharply highlight the growing view that there is heterogeneity in the mechanisms producing longitudinal inflammatory lesions, some of which may be independent of aquoporin-4 autoimmunity. The need to distinguish multiple sclerosis from neuromyelitis optica The importance of separating NMO from MS is twofold. Firstly, NMO has a worse outcome than MS, with frequent and early relapses. Within five years of onset, 50% of patients are blind in both eyes and cannot walk unassisted, and 20% die of respiratory failure due to cervical myelitis. Secondly, NMO responds to immunosuppressive therapy with agents such as azathioprine and rituximab, [61],[62] whereas the currently promoted treatment of MS includes immune-modulating agents such as interferon. Optic spinal form of MS cases in Japan which were seronegative for NMO-IgG responded well to interferon even when there was concomitant LETM. [42] Indian OSMS is most likely to be closer to conventional MS than NMO and therefore should be given the benefit of treatment with disease modifying agents such as beta interferon. Conclusion A relatively familiar condition, ATM has become transformed with recent developments, especially the advent of the MRI and the discovery of biomarker NMO-IgG. It is the clinician's responsibility to label ATM according to internationally established criteria, starting with a clinical description of partial or complete TM. Imaging studies should be done in the acute phase of the illness so as to accurately measure the length of spinal cord lesions and should include the brain as well. Reasonable investigations should be done to establish the idiopathic nature of ATM. A long-term follow-up study helps to reasonably ascertain conversion to MS or an NMO phenotype disorder. In the Indian context, some of our recent studies have highlighted the benign nature of Indian OSMS and distinguished it from the aggressive and disabling Japanese form. Longitudinal spinal cord lesions are not necessarily associated with a bad prognosis, especially in Indian males with an NMO spectrum disorder. The NMO seronegative nature of majority of Indian patients could indicate that these disorders may have autoimmunity against a nonaquaporin target. Recent detection of novel biomarkers for paraneoplastic ATM associated with LETM, is a good example for nonaquaporin autoimmune disorders. Additionally genetic heterogeniety may be responsible. Japanese OSMS is strongly associated with MHC Class11 allele DPB1FNx01 0501, [10] while Class 11 allele DRB1FNx01 1501 is linked to Western form of MS. Prior to the detection of NMO-IgG antibody, opinions differed as to whether MS and NMO were the same or distinct disorders. However, the observations that NMO-IgG is not found in all cases of NMO or NMO spectrum disorders and that about 10% of classical MS patients also carry the antibody, erodes its credibility. The question that needs to be answered once again is whether, based on this biomarker, we should reclassify CNS demyelinating disorders into MS and autoimmune aquaporinopathies. Is NMO-IgG responsible for disease causation or is it simply an epiphenomenon? References
Copyright 2009 - Neurology India The following images related to this document are available:Photo images[ni09039f1.jpg] [ni09039t1.jpg] [ni09039f2.jpg] [ni09039f3.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}