 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
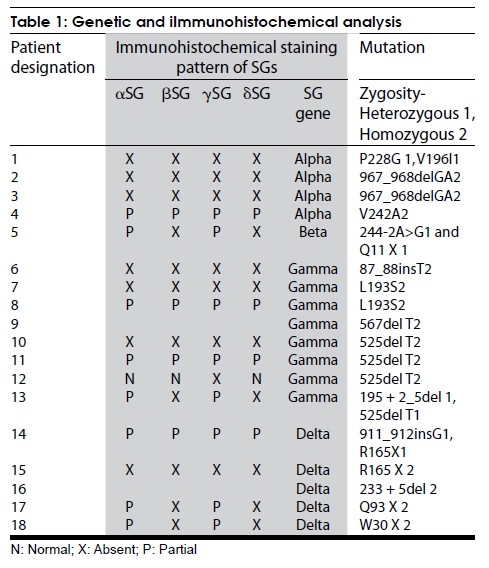
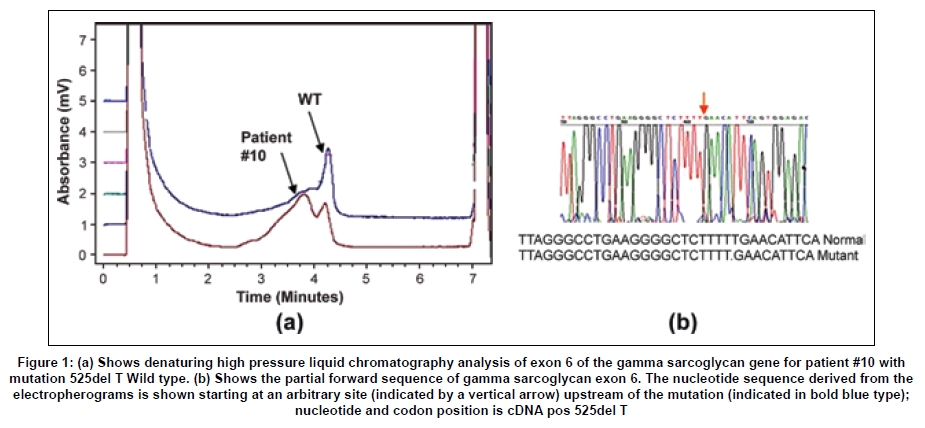
Neurology India, Vol. 57, No. 4, July-August, 2009, pp. 406-410 Original Article Spectrum of mutations in sarcoglycan genes in the Mumbai region of western India: High prevalence of 525del T Satish V. Khadilkar, Rakesh K. Singh1, Madhuri Hegde2, Andoni Urtizberea3, Don R. Love4, Belinda Chong2 Departments of Neurology, Grant Medical College and Sir JJ Group of Hospitals, Correspondence Address: Dr. S.V. Khadilkar, 110, New Wing, First Floor, Bombay Hospital, 12 New Marine Lines, Mumbai, India. khadilkar@vsnl.com Date of Acceptance: 18-Feb-2009 Code Number: ni09118 PMID: 19770540 DOI: 10.4103/0028-3886.55603 Abstract Background : While the clinical and immunocytochemical features of sarcoglycanopathies have been reported from India, genetic aspects have not been studied. There is large variation in the sarcoglycan mutations among the studied populations. Keywords: Genotype, sarcoglycanopathies, western India, 525del T Introduction Sarcoglycanopathies manifest with progressive weakness of the limb girdle muscles presenting in childhood or adult life. These muscular dystrophies result from mutations in one of the four sarcoglycan genes [SG].[1] Studies of different populations have revealed a variable frequency of sarcoglycanopathies as a clinical subgroup of neuromuscular diseases, as well as variable frequencies of mutations in each of the four SGs. [2],[3] The phenotypic and immunocytochemical analysis of Indian sarcoglycanopathy patients have been described earlier in small hospital-based studies. [4],[5],[6] No information is available on the genetic aspects of Indian patients having sarcoglycanopathies. Such information is relevant to establish local prevalence data. Materials and Methods This study was carried out at the neuromuscular clinic of a tertiary Neurology referral center. Informed consent forms in the languages understood by the patients were obtained and the study was approved by the hospital board. The study period lasted 6 years ending in December 2000. Patients of either gender with chronic progressive weakness of limb girdle muscles fulfilling Bushby's criteria were included. [7] Detailed pedigree charting was done and all available family members were examined. All patients provided a detailed medical history and underwent a clinical neurological examination. A total of 32 pairs of muscle were tested in each patient. The weakness of all groups of muscle was graded on a 0-10 point system from which an average muscle score (AMS) was derived. [8] Serum creatine kinase (CK) levels were determined at the initial presentation. Electromyography and nerve conduction studies were performed using the Dantec system. Electrocardiography was done on all patients and echocardiography was performed when feasible. Depending upon the clinical involvement, quadriceps, biceps, or adductor magnus muscles were biopsied. Muscle biopsies were studied for histology, histochemistry, and immunocytochemistry for all four sarcoglycan proteins (Novocastra laboratories). Sarcoglycan gene mutation analysis was performed as described by Hegde, et al . [9] using denaturing high pressure liquid chromatography (DHPLC) analysis and direct sequencing. The complete coding region of the four sarcoglycan genes, including all splice junctions, was amplified in a total of 32 fragments. The sequence of the primer pairs, their corresponding amplicon sizes and optimized conditions for polymerase chain reaction (PCR) amplification and DHPLC analysis can be found at www.dmd.nl/SGC_DHPLC.html. All primers were checked against the NCBI-SNP database ( http://www.ncbi.nlm.nih.gov/SNP/ ) and the HGV base ( http://hgvbase.cgb.ki.se/ ) to avoid overlapping with single-nucleotide polymorphisms (SNPs). In the case of the subsequent sequencing of amplicons, products were purified using the Concert Rapid PCR Purification System (Invitrogen), which was subjected to cycle sequencing using the BigDyeTM Terminator Cycle Sequencing, Version 2 kit (PE Applied Biosystems, Foster City, CA). Sequence variants were called mutations only if they have been previously described in literature as mutations ( www.leiden.nl ). Results Out of the 68 patients [probands] studied completely with immunohistochemistry and genetic tests for sarcoglycanopathies, 18 patients showed mutations in the SG gene [26.4%]. There were 9 males and 9 females. Family history was positive in 6 patients and 6 patients were born of consanguineous marriages. The mean age at presentation was 22.5 years old (range: 10-45 years old), the mean age at the onset of the disease was 15.7 years old (range: 3-30 years old), and the mean duration of the disease was 6.4 years (3 months-20 years). [Table - 1]a and b show the frequency of affectation and the severity of weakness of the various muscles. The mean CK value was 1286 IU; this value was higher with the shorter duration of disease. A muscle biopsy was performed on 16 patients; 2 patients refused a biopsy. Histological analysis confirmed the clinical diagnosis of muscular dystrophy in all the patients, as was evident by variation in fiber size, degeneration and regeneration of fibers, and the proliferation of connective tissue. Immunocytochemistry showed the complete absence of all four SGs in 7 patients and 4 patients had a partial absence of immunostaining for all SGs. One patient showed the absence of only gamma SG staining, while the other 3 patients with SGs exhibited normal staining. In the remaining 4 patients, beta and delta SG staining was completely absent and alpha and gamma subunits were partially affected [Table - 1]. Disease causing mutations were documented in the alpha SG gene in 4 patients, in the beta SG gene in 1 patient, in the gamma SG gene in 8 patients, and in the delta SG gene in the remaining 5 patients [Table - 1]. The most common gene abnormality was the 525del T in the gamma SG gene [Figure - 1]. An electrocardiogram was performed on all patients, but no abnormality of rhythm was detected in any patient. A 2 D echocardiography was performed in 12 patients and was abnormal in 5 patients, suggesting reduced contractility of the myocardium and segmental wall motion abnormalities. Mutation nomenclature as described by "den Dunnen JT, Antonarakis SE. Nomenclature for the description of human sequence variations. Hum Genet. 2001 Jul; 109:121-124. Mutation database reference: Leiden Muscular Dystrophy Database ( www.dmd.nl ), Human Genome Mutation Database ( http://www.hgmd.cf.ac.uk/ ) Discussion The proportion of sarcoglycanopathies among patients with limb girdle muscular dystrophy (LGMD) is known to vary according to the studied population. In Brazil, the relative proportion for the sarcoglycanopathy group was found to be 32%, which is among the highest figures. [10] In the Italian population, sarcoglycanopathy constituted 18.1% of the population, [11] the North of England population constituted 11.7%, the United States population constituted 15%, [12] and in he Netherlands, sarcoglycanopathies account for 16% of the classified families. [13] In this study, sarcoglycanopathies constituted 26.4% of patients with LGMD. This figure from western India is higher than most of the studies quoted above, suggesting that sarcoglycanopathies are more common in the population of western Indian. However, further studies including other parts of India will be required to know more about the prevalence of sarcoglycanopathies in India. In this study, alpha, beta, gamma, and delta SG mutation frequency was noted to be 4:1:8:5. While this is different from studies of out-bred populations wherein the relative frequency is noted to be in the proportion of 8:4:2:1, [2] wide regional variations of the relative subgroup frequencies are well-known. In a recent paper from France, the proportion was 55.3% alpha, 25.5% gamma, 17% beta, and only 2.2% had delta SG mutations. [14] However, in the Tunisian population, gamma sarcoglycanopathies are more common [15] than other types, a finding similar to western India. The 525del T was encountered in 50% of the gamma SG mutations, three homozygous and one heterozygous form. The occurrence of this deletion in the Indian patients is of much interest, as it has been known to occur mainly in select inbred populations. [16] This finding may suggest this deletion to be a hot spot in the gene or conceivably, may have an explanation in the human migration patterns. Further work on the haplotyping of these patients will shed more light on this issue. The absence of c 283y, the gypsy mutation, [17] is also of note as the gypsies are believed to have originated in India. None of our patients had intellectual or cardiac symptoms, but echocardiogram analysis showed reduced ejection fraction in 2 of the 8 patients, suggesting subclinical cardiac involvement. Of the 4 patients with alpha SG gene mutations, 2 had severe phenotypes and 2 had milder phenotypes. The gene mutations were different in each of them, as is expected in an out-bred population. The R77C mutation described largely in inbred populations was not detected in the study described here. The single patient with a beta SG mutation had a strong family history with five affected members in two generations. His disease started at 8 years of age, but at 12 years of age he was severely disabled, requiring a wheelchair for ambulation. He had echocardiographic evidence of cardiomyopathy, which was clinically asymptomatic. Delta SG mutations are uncommon and reported infrequently in most populations, [1] but are common in Brazil. [18,19] Our study included 5 patients with delta sarcoglycanopathy, but while this represents one-third of all our patients analyzed at the genomic level, a larger study would be required to estimate the true prevalence of delta SG mutations in the Indian population. As expected, a majority of the patients had proximal limb girdle muscle weakness. The hip girdle was affected more severely than the shoulder girdle and there was preferential weakness of hip adductors and hamstring muscles. In the upper limbs, the biceps and deltoids were the weaker muscles. This pattern of proximal muscle weakness was similar to that seen in other studies, [1],[20],[21],[22],[23],[24] including our previous study. [4] The clinical profile of various mutations was indistinguishable from each other. The most common pattern of staining was of total or partial absence of all four SG proteins [11/16]. This is in keeping with Mizuno's hypothesis of secondary loss of membrane SG proteins. [25] Studies of the relationship between the severity of clinical expression and the extent of loss of SG proteins have provided data in favor, as well as against, a positive correlation. [26],[27],[28] In this study, in general, the extent of SG staining abnormalities did not match completely with the clinical severity or the duration of the disease. [19] A correlation was found in patients with a delta sarcoglycanopathy. Of the four biopsies performed on patients with delta sarcoglycanopathy, those three with partial protein loss had the milder phenotype. One of them was heterozygote at the molecular level. The second pattern of absence of beta and delta SG proteins is intriguing. In these patients, the alpha and gamma SG proteins were only mildly reduced in intensity. Studies of the sarcoglycan complex have suggested that beta and delta sarcoglycans are the most tightly associated and form a functional core of the complex.[29] The specific assembly pathway of sarcoglycans is dependent on beta and delta sarcoglycans.[30] Chan, et al .[29] postulated that beta, gamma, and delta sarcoglycans may function as a receptor for an extracellular ligand, whereas alpha sarcoglycan acts as a downstream effector. The conclusion from this study also argues for a functional interaction between beta and delta SG, but the significance of this conclusion is based on a small number of patients. In the present series, 1 patient with a del525 T gamma SG mutation exhibited the isolated absence of gamma sarcoglycan; the other three SGs stained normally. This rare occurrence has been reported previously in only two case reports. These reports also emphasized the fact that a severe phenotype could result from the isolated loss of one SG protein, adding complexity to the issues of genotype to phenotype correlation. [31],[32] As alpha sarcoglyanopathies are more frequent in some studied populations, it has been suggested that the search for sarcoglycanopathies should screen initially for aberrant alpha sarcoglycan. This approach may not be applicable in all populations; hence the routine unbiased analysis of all four proteins should be considered mandatory, [1] as the isolated use of the alpha SG antibody would have led to a misdiagnosis of some of our patients. Conclusions Sarcoglycanopathies constituted 26.4% of limb girdle muscular dystrophies in the Mumbai region in Western India. Mutations in the gamma and delta SGs were more common than the alpha and beta SG genes. The interesting occurrence of 525del T is noted, but needs further elucidation. The clinical profile of various mutations was indistinguishable from each other. Immunocytochemical patterns highlighted the importance of using all four antibodies. Acknowledgements We wish to thank the muscular dystrophy society of Mumbai and all our patients who participated in this study. Special thanks are due to late Dr. Anil D. Desai for his encouragement. References
Copyright 2009 - Neurology India The following images related to this document are available:Photo images[ni09118t1.jpg] [ni09118f1.jpg] |
| |||||||||


{kind=link}
{kind=link}