 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 57, No. 4, July-August, 2009, pp. 460-463 Brief Report Pyridoxine-dependent seizures: 10-year follow-up of eight cases Roshan Koul Department of Child Health, College of Medicine and Health Sciences, Sultan Qaboos University Hospital, Muscat, Oman Correspondence Address: Dr. Roshan Koul, Senior Consultant and Child Neurologist, SQUH. BW1, Muscat, Oman. roshankoul@hotmail.com Date of Acceptance: 16-Feb-2009
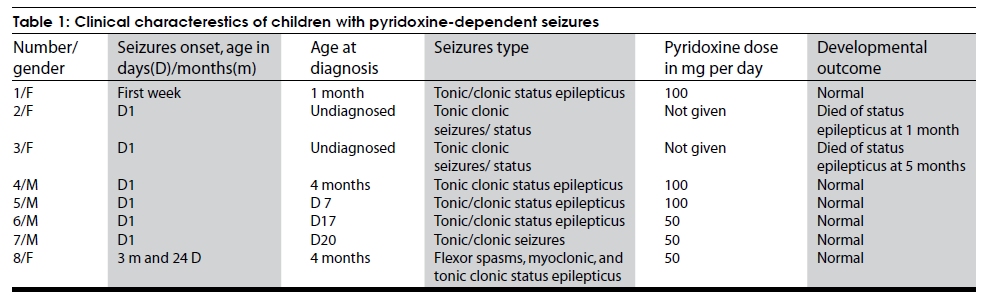
Code Number: ni09128 PMID: 19770548 DOI: 10.4103/0028-3886.55595 Abstract Eight children with pyridoxine-dependent seizures (PDS) were seen over a period of 10 years. Of those children, 6 are on regular follow-up. Four of the children were seen in one family. All the patients presented with refractory seizures, mainly neonatal status epilepticus. Though PDS is a rare condition, it must be considered in all cases with refractory seizures, particularly in childrens younger than 3 years. When confirming a diagnosis, oral pyridoxine is as effective as intravenous pyridoxine.Keywords: Pyridoxine, pyridoxine dependency, neonatal status epilepticus, refractory seizures Introduction Refractory seizures in neonates and in early infancy is a major therapeutic challenge. Many of these are syndromic seizures and are resistant to treatment. Pyridoxine-dependent seizures can wrongly form a refractory seizure group if they are not diagnosed. However, response to pyridoxine treatment is dramatic in a child previously labeled with refractory seizures. A report of a series of eight cases is described from Oman. Materials and Methods All children aged less than 1 year with refractory seizures were admitted to the pediatric intensive care unit (PICU) for seizure treatment. When no definite etiology could be established and a neurological examination including investigations were s normal, a trial of pyridoxine was given. Baseline investigations such as CBC, liver function tests, bone profile, lactate, tandem mass spectrometry, electroencephalograph (EEG), and imaging were done for all patients. Pyridoxine dependency was diagnosed when there was a prompt response of clinical symptoms (seizures) to the administration of the pharmacologic dose of pyridoxine (100mg per day). Results Of the eight children seen, four belong to one family. The age at the time of diagnosis of pyridoxine dependent seizures was Day 7 to 4 months (mean 30 days). It was earliest in the familial cases. Six of the children had seizure onset on Day 1 of life. All the children presented with status epilepticus not responding to antiepileptic drug treatment. Only the first case was given intravenous pyridoxine (100mg) and all other children received oral pyridoxine, 50mg 3 children and 100mg 2 children. The child who received intravenous pyridomine had hypotension, bradycardia, and apnea needing resuscitation. In all the children seizures stopped immediately after pyridoxine and the subsequent development was normal [Table - 1]. The clinical characteristics and the management at the other facilities before admission to this hospital of the eight children were: Case 1 A 21-day-old female child had tonic-clonic seizures in the first week of life. She was started on phenobarbital and as the seizures were not controlled, phenytoin sodium and clonazepam were added. After 3 weeks of uncontrolled seizures, the patient was transferred to our hospital. A trial of pyridoxine 100 mg was given intravenously in the ICU. The seizures stopped completely. This child had hypotension, bradycardia, and apnea, which needed resuscitation. The patient has been seizure free for 9 years now and is developing normally. Cases 2 to 3 Both the children belong to the same family of four children with pyridoxine dependent seizures (Cases 2-5) and in both the children seizures started in the first week of life. Both the children died, Case 2 at 1 month of age and Case 3 at 5 months of age, due to seizures and status epilepticus not recognized as pyridoxine dependent seizures. Case 4 Male child aged 4 months was referred for neonatal onset tonic-clonic and myoclonic seizures and status epilepticus refractory to treatment, phenytoin, phenobarbital, and midazolam infusion. His EEG showed multifocal seizures. A trial of oral pyridoxine (50mg twice a day) was given on the third day of admission to our institute. Seizures did not recur after 3h of pyridoxine administration. He has been seizure free for the last 4.5 years and his development was normal. Case 5 A male child presented with tonic-clonic seizures on Day 7 and the seizures responded well to pyridoxine. He had normal development. Case 6 A 15-day male child developed tonic-clonic seizures on Day 1. He was started on phenobarbital, as the seizures were not controlled phenytoin loading was done and subsequently clonazepam was added to the treatment regimen. As the seizures continued to recur, he was started on midazolam infusion. At this stage the child was transferred to our institute. On Day 17 of birth (on the second day of admission) he was given a trial of oral pyridoxine (50mg) and seizures stopped within three hours. He has been seizure free since three and half years and his development was normal. Case 7 A 18-day old male child developed tonic-clonic seizures on Day 1 of birth. He was put on phenobarbital at the other facility. After the discharge he had recurrence of seizures for which he was treated with oral pyridoxine with which he had good control of seizures. He has been seizure free for the last two years and his development was normal. Case 8 A 3-month and 24-day old female child was admitted to the Pediatric Intensive Care Unit with tonic-clonic status epilepticus refractory to phenytoin, phenobarbital, and midazolam infusion. Her seizures started as tonic-clonic seizures, later myoclonic and even flexor spasms were added. In the PICU a trial of 50mg pyridoxine was tried. There was no seizure recurrence within the next 24 hours. She has been seizures free for the last two year and the development was normal. Discussion Since the first descriptin in 1954, most of the reports worldwide on pyridoxine-dependent seizures (PDS) were either single case or few case series. [1] PDS should be considered as a possibility in childhood refractory seizures particularly under 3 years of age with no obvious etiology. [2] Seizure semiology can be of any type including infantile spasms. In the majority, the sezures manifest in the neonatal period or early infancy, however seizures can occur for the first time up to the age of three years. [2],[3] The reported mean age at the time of diagnosis was 3.5 months, range 1 day to 32 months, The diagnosis was earlier in the familial cases [4] In this series the age at the time diagnosis was Day 7 to 4 months (mean 1 month). Worldwide more than 100 cases have been reported.[4] In the United Kingdom epidemiological study of pyridoxine-dependent and pyridoxine-responsive seizures, the point prevalence of definite and probable cases in children under 16 years of age was 1 per 687000 and the birth incidence was 1/78300. [5] Only two cases have been reported from Saudi Arabia. [6] The diagnosis of PDS is established by intravenous or oral treatment pyridoxine trail. The criteria include: (1) cessation of clinical and electrographic seizures following intravenous administration of 100mg of pyridoxine while the patient is being monitored with EEG and (2) no seizure recurrence while on long-term pyridoxine treatment. There have been several reports of bradycardia, apnea, and hypotension when given intravenously. [6],[7] We also observed this side effect when using pyridoxine intravenously in the first case. Whether given intravenously or orally, pyridoxine doses are equally effective. The response to oral administration occurs in no more than 12 hours, and intravenous injection should be restricted to patients with status epilepticus cases only. [7] After the experience with the first case, we use only oral pyridoxine 50mg twice daily for 2 days as therapeutic trial. If the seizures do not stop, a diagnosis of PDS is excluded. In our observation, all patients with PDS responded within 24 hours when receiving 50mg twice a day. In fact, no seizures occurred in responders after a few hours of the first dose of pyridoxine, though there are reports that oral administration of 15mg/kg may take up to 1 week to stop seizures. [7] A daily dose of 15-300mg/kg has been recommended for lifelong treatment in these patients. Our experience showed that a dose of 5mg/kg to 20mg/kg/day was enough to start. We have not increased the dose since the start of treatment. Seizures of various types such as infantile spasms, myoclonia, partial seizures, and generalized seizures have been seen in patients with PDS. [7] We have encountered only generalized tonic clonic seizures and all of them had refractory status. Two of the children had myoclonic seizures and one child had infantile spasms as well. An EEG pattern of suppression burst, polymorphous tracings, spikes, polyspikes, high voltage rhythmic delta slowing, and hypsarrhythmia have all been seen in these children. [7] An initial genetic study in five families from the U.S.A. mapped a locus for PDS at chromosome 5q31; [8] however, another study excluded assignment to this locus alone, indicating genetic heterogeneity for pyridoxine-dependent seizures. [9] Alpha aminodipisemialdehyde (ALDH7A1) is the only gene known to be associated with PDS. [10] No genetic study was carried out in our cases. Six of the 8 children are on follow-up; the first one has been on follow-up for over ten years. All of these children are developing normally. If started early, pyridoxine treatment prevents mental retardation and development is normal. [11] If PDS is diagnosed late in childhood, there is significant developmental delay and mental retardation seen in almost 60% of the cases.[12] A different phenotype has been reported recently, in them the pyridoxine treatment did not affect the outcome and all had severe long-term deficits and mental retardation.[13] Another resistant form of epilepsy has been described recently, which did not respond to pyridoxine treatment, but responded to pyridoxal phosphate. This has been labeled as pyridoxal phosphate-dependent neonatal epileptic encephalopathy. In this condition, seizures do not respond to pyridoxine or antiepileptic drugs but only to pyridoxal phosphate administration. [14] Lately, PDS has been described under neurotransmitter disorders.[15] Adults and adolescents need approximately 1 to 2mg per day, while infants and young children require 0.3 to 0.6mg per day. [16] Pyridoxine dependency is diagnosed when there is a prompt response of clinical symptoms to the administration of the pharmacologic amounts of pyridoxine. Life-long administration of pharmacologic amounts (5-300g/kg/day) is necessary to prevent the recurrence of convulsions. [4] In the U.S.A., a patient registry called the "pyridoxine-dependent seizures patient registry" has been made available. All the patients diagnosed in the United States and Canada are filtered through the Children's Hospital and Regional Medical Center in Seattle, WA. The registry can be contacted through the author, sgospe@u.washington.edu, or at pyridoxine@seattlechildrens.org. In conclusion, we describe follow-up of six children with PDS. All of the patients presented with refractory status epilepticus, mostly with tonic clonic seizures. All are developing normally. One has to consider this condition in any drug-resistant epilepsy in infancy, when no underlying cause is found. Oral pyridoxine is as good as intravenous for confirmation of diagnosis. References
Copyright 2009 - Neurology India The following images related to this document are available:Photo images[ni09128t1.jpg] |
| |||||||||

{kind=link}