 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
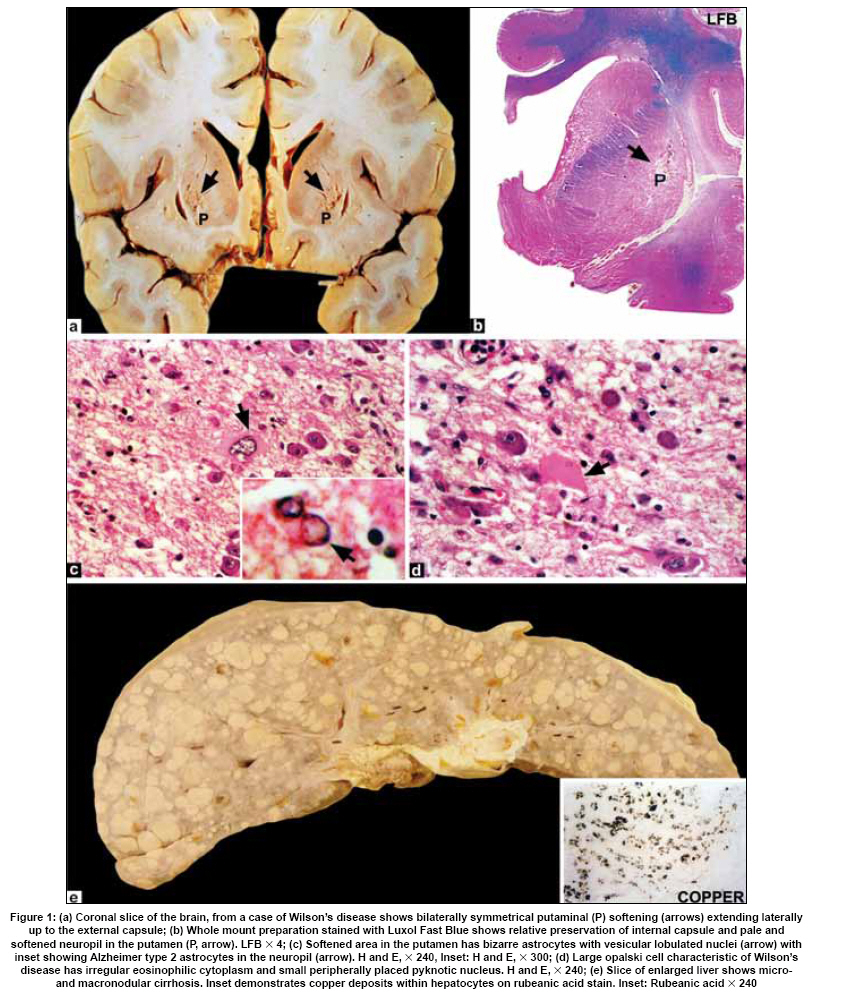
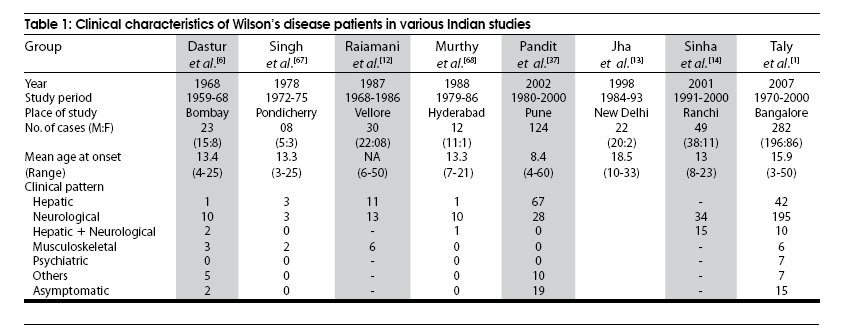
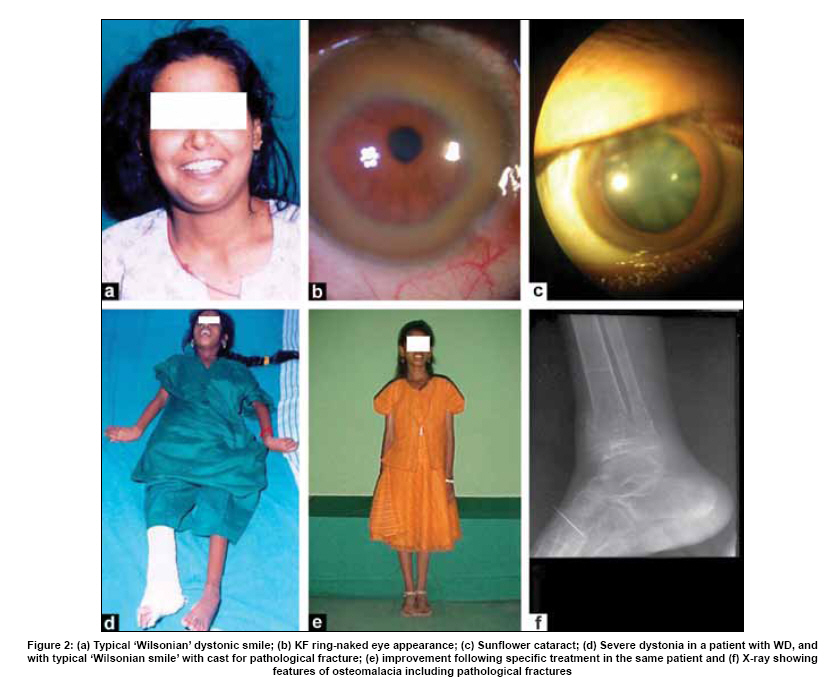
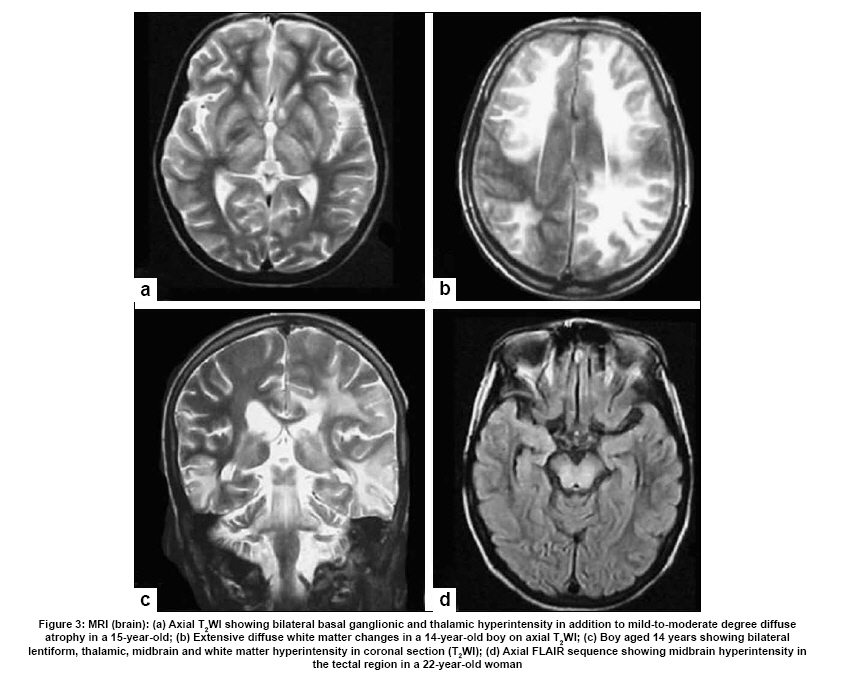
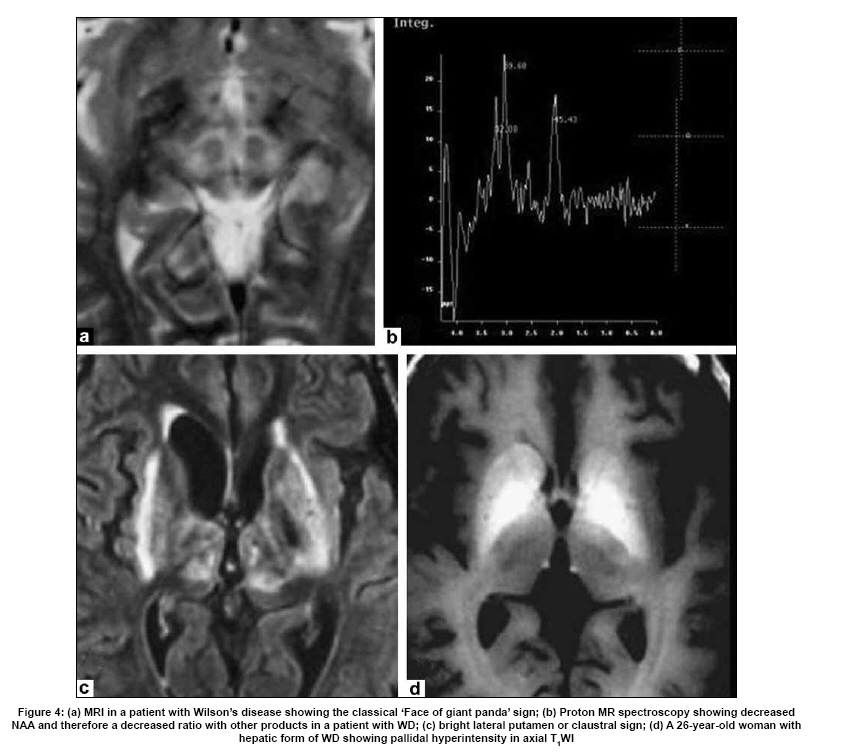
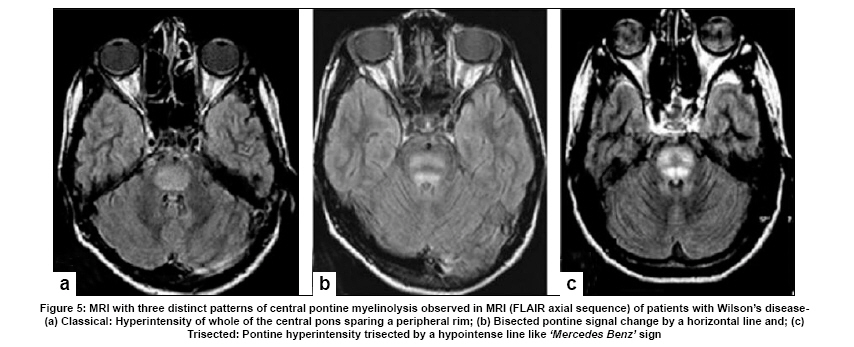
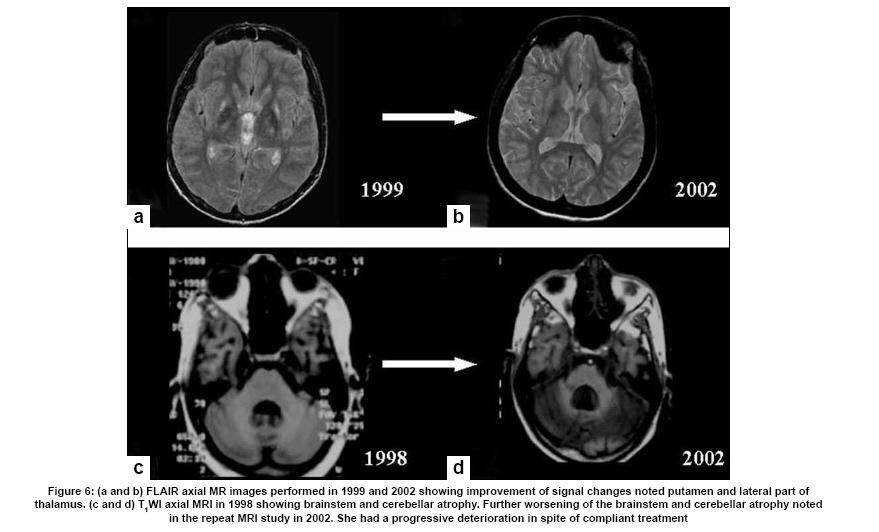
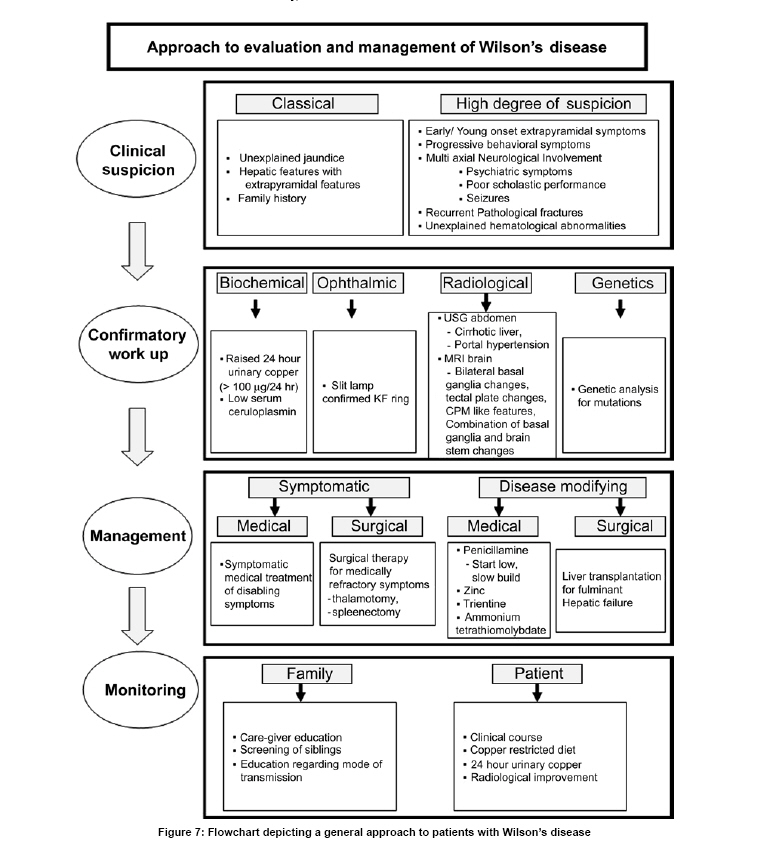
Neurology India, Vol. 57, No. 5, September-October, 2009, pp. 528-540 Indian Perspective Wilson's disease: An Indian perspective A. B. Taly, L. K. Prashanth, S. Sinha Department of Neurology, NIMHANS, Bangalore, India Date of Acceptance: 06-Oct-2009 Code Number: ni09161 PMID: 19934550 DOI: 10.4103/0028-3886.57789 Abstract Wilson's disease (WD) is an autosomal recessive disease involving a defect of copper transport by the hepatic lysosomes. It leads to excess copper deposition in the liver, the brain, the kidneys and the skeletal system, affecting most commonly children or young adults and running an invariably fatal course if not adequately treated by de-coppering therapy. The last century has witnessed several changes, notable among these are: Increased awareness, improved diagnostic facilities leading to earlier recognition even in the pre-symptomatic phase, clear distinction from its mimics, aggressive therapeutic approaches owing to availability of effective treatment and an overall reduction in the morbidity and mortality. It is widely acknowledged that the disease is not as rare as once believed. Sir SAK Wilson published his landmark article in 1912, but it was only in 1968 that the first patient of WD was reported from our country. Publications from India on WD have focused on phenotypic characterization, documentations of lesser recognized aspects of the disease e.g. seizures, behavior abnormality, speech and cognitive impairment, sub-clinical affection of visual pathway, heart and autonomic function and pre-symptomatic detection. Attempts have been made to understand the clinical heterogeneity of the disease through identification of biochemical and immunological markers, magnetic resonance imaging, neuropathological study and genetic analysis for novel and/or known mutations. Assessment of impairment and severity and effect of various therapeutic interventions namely zinc sulphate on the long-term outcome and quality of life have also been studied. Nevertheless, clinicians often face difficulties in long-term care of these patients. Diagnostic errors leading to delay in diagnosis and initiation of treatment are common, even in patients with positive family history. There is no consensus regarding therapeutic protocols since the use of penicillamine, once a 'gold standard' for treatment, has been debated by experts. Mortality and morbidity of this potentially treatable disease and nonavailability of medications to the poor patients remain a major area of concern.Keywords: India, Wilson's disease, MRI, pathology Introduction In the last few decades genetically inherited disorders have gained importance, primarily due to improved genetic and biochemical testing. An increasing emphasis of research is on the therapeutic aspects of these disorders. Among the inborn errors of metabolism, Wilson's disease (WD) is of specific interest to clinicians as it is potentially curable disease if recognized and treated early. Wilson's disease like many other inherited metabolic diseases has geographical variation in the epidemiology, clinical presentation and course. In the late 1960s, WD was considered as non-existent in India. However, over the years there have been many publications describing diverse and unique manifestations of the disease in this part of the world and including perhaps the largest series in the world literature. [1] History Recognition of WD was credited to S.A. Kinnier Wilson who in his classic paper 'Progressive lenticular degeneration: a0 familial nervous disease associated with cirrhosis of the liver' in 1912 documented a detailed description of clinical and pathological changes in WD, which still holds well till date. [2],[3],[4] However it took more than a half century to document first series of patients with WD in India. [5] The credit for the first detailed description of clinical, biochemical and pathogenetic aspects of WD in India goes to Dastur and Manghani, who reported various aspects of WD in 23 patients from 16 families. [6] Subsequently several other centers from the country have shared their experience. Notable among them is the contribution from National Institute of Mental Health and Neurological Sciences (NIMHANS), Bangalore in south India. [1] Professor H. Sathyanarayana Swamy started a specialty clinic for WD at NIMHANS in 1970, which has become a major referral and research center for WD in south India. The clinic provides care mainly to the neuropsychiatric form of WD. However, patients with non-neurological forms and in pre-symptomatic stage are also evaluated for the confirmation of diagnosis and are on long-term follow-up. Most of the earlier reports from the country have focused on clinical and therapeutic aspects of the disease, the genetic studies have started in the early 2000s. [7] Epidemiology The reported prevalence of WD varied between 12 and 29 per 100 000 in European population. [8] The prevalence in Asian countries other than India varied between 33 and 68 per 100 000. [9] There are no community-based incidence and prevalence studies of WD in India. Most of the studies were hospital-based. In the study of hepatobiliary spectrum disorders from Lucknow, north India, of the 235 patients studied over a period of 3, WD accounted for 18 patients (7.6%). [10] A large multicenter study, Pediatric Liver Study Group of India, in 11 medical colleges across the country studied the etiological spectrum of chronic liver disease in pediatric age group using a structured questionnaire. Of the 948 children with chronic liver disease, metabolic liver diseases accounted for 194 (19.7%) patients. Wilson's disease was the most common metabolic liver disease in this study; however, the exact frequency has not been provided. [11] Recent Indian literature has focused on neuropsychiatric form of WD. [1],[12],[13],[14] At WD specialty clinic in NIMHANS, about 15-20 new cases of WD are registered annually. Genetics Wilson's disease is an autosomal recessive disorder of hepatocyte copper trafficking caused by impaired function of P-type adenosine triphosphatase (ATPase), encoded by ATP7B gene located on chromosome 13q14 and consists of 21 exons. This important genetic breakthrough came in 1993 from three independent groups. [15],[16],[17] Until now more than 400 mutations in the gene have been documented from various countries. [18] Genetic analyses from India have reported mainly from three centers: Chandigarh, Kolkata and Vellore. [19],[20],[21] The commonest mutations in these studies are variable and include: (a) Chandigarh group: T3305C, C2975A, 2977insA and 3031insC-6% each; (B) Kolkata group: C813A-19% and (c) Vellore group: G3182A-16% and C813A-12%. Till date all together, a total of 51 mutations of ATP7B have been documented in India including 34 novel mutations (Chandigarh-18, Kolkata-5, and Vellore-11). Of the mutations documented in India, C813A is the common mutation. There is no single predominant mutation noted in the Indian population unlike the studies in other countries: PH1069Q in 60% of central European population and pR778L in 45% of Chinese population, thus suggesting genetic heterogeneity in India. [18] Pathogenesis and Pathology The exact pathophysiology of WD was uncertain till 1950s, despite a good descriptive account of clinical presentation and documentation of excess of copper in liver and cornea. The major breakthrough came in 1948, when Cumings demonstrated excess of copper in both brain and liver in patients with WD and suggested de- coppering with British antilewisite (BAL). Subsequent to this work, genetic studies demonstrated that mutations in ATP7B, a copper transporting protein leads to abnormal accumulation of copper in hepatocytes, which later spills into the circulation resulting in deposition of copper in various body organs. Copper, a trace element forms an active center of the redox copper enzymes in copper-zinc superoxide dismutase, ceruloplasmin and cytochrome oxidase. It is hypothesized that excessive copper deposited in various tissues leads to free radical damage. Sinha et al., [22] estimated the serum malondialdehyde (MDA), a standard test to detect such oxidative damage, and detected increased levels of MDA in serum in 50% of patients. The same workers have also assessed the serum tocopherol level, a naturally occurring antioxidant that might protect tissues from oxygen-derived free radicals among 34 patients and noted reduced levels of tocopherols in 59% of patients. However, serum tocopherol levels did not correlate with the clinical or biochemical status. [23] In another study to ascertain the role of various cytokines (IL-2, IL-4, IL-6, IFN-γ, TNF-α) in patients with WD, it was noted that both the pro- and antiinflammatory cytokines were significantly elevated in patients when compared to controls. However, the cause and effect relationship of elevated cytokines to the ongoing process in WD could not be to ascertained. [24] Thus, the mechanisms whereby copper deposition leads to tissue damage remains largely unanswered. There is paucity of literature on the clinicopathological correlation in WD in the international literature. In an autopsy study of eight patients of WD, the pathology described included: Central pontine myelinolysis like changes, white matter cavitations, putaminal softening, ventricular dilatation and atrophy. The authors emphasized that the lenticular involvement in WD was not universal as believed, and pathological involvement was far more diffuse in character [Figure - 1]. [25] Clinical Profile The clinical presentations of WD are protean and varied. Walshe has aptly stated that no two patients of WD may have similar clinical characteristics even among the common sib-ship, stressing its diverse manifestations. [26] The phenotypic variability of WD often leads to delay in diagnosis, unless there is a high index of suspicion. In a large series of 307 patients of WD, evaluated at NIMHANS, Bangalore, misdiagnosis at initial evaluation was recorded in 62.5% cases, leading to a mean delay of two years in arriving at correct diagnosis and initiating treatment. [27] The literature regarding WD from India was non-existent till 1960s, when a few case series have been reported. [5],[28] Following the initial article by Dastur et al., [5] WD has been more frequently recognized in our country. [6],[29] Dastur and Wadia pointed out that in the Indian subcontinent, musculoskeletal form might be more common. [5] The salient demographic profile and clinical types of WD across the country has been summarized in [Table - 1]. The notable difference was the rarity of musculoskeletal form of WD in other series from India. The presenting symptoms in different series have been variable. In a large series of 307 patients from NIMHANS, the notable clinical features were: Tremors (31.6%), dysarthria (15.6%), jaundice (12.4%), abnormal gait (8.8%), abdominal distention (7.8%), musculoskeletal symptoms (5.2%), seizures (4.9%), behavioral problems (4.6%), dystonia (3.6%), clumsiness (2.6%), drooling of saliva (2.6%), generalized weakness (2.3%), poor scholastic performance (1.9%), altered sensorium (1.3%), bleeding diathesis (1.3%), dysphagia (0.9%), chorea (0.3%) and poor vision (0.3%) [Figure - 2]. [27] Though hepatic involvement occurs early in the course of WD, it may go unrecognized and may not be the first symptom for seeking clinical attention. In the series by Walshe et al., [30] hepatic presentation was the major presenting symptom in approximately 40% of patients followed by neurological manifestations in 40% of patients. However, in other series, neurological manifestations were the presenting feature in about 60% of patients. [31] Extrapyramidal manifestations are the dominant neurological features; [1] however, seizures, cognitive disturbances, [32] behavioral problems [33],[34] and recurrent limb weakness [35] have also been reported. Hepatic presentation is predominantly noted at younger age. Most of these patients present with jaundice but do not get investigations for WD because of low index of suspicion and also rarity of Wilson's disease in the community. Hepatic involvement ranges from acute fulminant hepatitis to subclinical chronic liver disease. [36],[37] Some of the patients with hepatic WD succumb to illness, while others later in the course of the illness develop neurological symptoms. [27] Psychiatric presentation, mostly case reports, of WD has also been recognized. [38],[39],[ 40] In a series of 350 patients with WD, in 15 patients psychiatric manifestations were the predominant symptom at the time of medical consultation. The psychiatric diagnosis was bipolar affective disorder, schizophrenia and psychosis with cognitive decline. [33] In a recent study of 12 patients, magnetic resonance imaging (MRI) and neuropsychological assessment suggested involvement of frontal-subcortical circuits in all but one patient. [41] In another prospective cross-sectional study of psychiatric disorders in 50 patients with WD, 24% of patients fulfilled the diagnostic criteria for syndromic psychiatric diagnosis. [34] Less common manifestations of WD include musculoskeletal [1],[6],[29],[42],[43] and hematological. [1],[44] Patients with WD are perhaps prone to develop malignancy like retinoblastoma, hepatocellular carcinoma, basal cell carcinoma and acute lymphoblastic leukemia. An interesting co- occurrence of glioblastoma multiforme and WD has been documented. [45] Diagnostics The laboratory diagnosis of WD can broadly be classified: Biochemical, ophthalmological, electrophysiological and radiological. Each of these investigations may have varied diagnostic and prognostication significance. Biochemical The key biochemical investigations in patients with WD include estimation of serum copper, serum ceruloplasmin and 24-h urinary copper. In addition to these, other biochemical investigations such as liver function tests, renal function tests and hematological investigations add to assess status of various other organs involved in the disease. In a study of copper metabolism in WD in north India, ceruloplasmin levels were significantly lower in patient population when compared to controls and there was a good co-relation between non-ceruloplasmin bound copper and 24-h urinary copper excretion. [46] Analysis of biochemical data in our patients in whom all the three parameters of copper metabolism (serum copper, ceruloplasmin and 24-h urinary copper) were available, revealed low serum ceruloplasmin in 88%, and increased 24-h urinary copper in 96% patients. When a combination either low ceruloplasmin or increased 24-h urinary copper was used, all but one of the 221 patients had abnormality (unpublished data). It should be noted that ceruloplasmin being an acute phase reactant might be falsely normal in patients with acute presentation especially with hepatic form. High index of suspicion in this group of patients is required for accurate diagnosis. These patients may also have involvement of other systems. Evidence of other target organ dysfunction in WD depends upon key clinical features, intensity of investigations and may vary with time. In a large series with predominant neuropsychiatric presentation, liver function tests were abnormally less than 10% of patients. [1] Ophthalmological evaluation Slit lamp evaluation for Kayser-Fleischer (KF) ring is an important diagnostic tests and forms corner stone of clinical diagnosis. The KF ring is invariably present in all patients with neuropsychiatric form of WD. The reported frequency of KF ring in various Indian series has been nearly the same and ranged from 86.6 to 97.1%. [1],[12],[13],[14] Its detection depends on experience of clinician and of the ophthalmologist when slit lamp examination is needed. In hepatic and other forms, KF ring may not be detected and therefore heavy reliance on this sign may lead to under-recognition of the disease. In a series of 282 patients, only 5 of the 37 patients with hepatic form had KF ring. [1] Sunflower cataract is infrequent and rarely symptomatic [47] [Figure - 2] b and c. Electrophysiological Electrophysiological studies have been done in WD to document any subclinical abnormalities and as such do not have diagnostic significance. Neurophysiological evaluation Only a few studies have reported electrophysiological studies such as electroencephalography (EEG), visual-evoked potentials, brain stem auditory evoked potentials, somatosensory-evoked potentials and nerve conduction studies. [1],[48],[49] In our study, the reported abnormalities included: EEG-41.1%, visual-evoked potentials-35% and brain stem auditory evoked potentials-42.1%. [1] Earlier Satishchandra et al., had found subclinical involvement of optic nerve and caudal brain stem auditory pathways. [48] In another study electrophysiological evaluation to study the visual pathway abnormalities in WD was carried out using electroretinography and visual-evoked potentials and has demonstrated reversibility of retinal dysfunction with clinical improvement. [50] Autonomic nervous system involvement has also been reported in WD. In a study, six of the 13 patients with electrophysiological evidence of dysautonomia had also clinical evidence of dysautonomia. It was concluded that subclinical dysautonomia in WD is perhaps due to central autonomic dysregulation involving both sympathetic and parasympathetic systems. [51] Abnormalities of cardiovascular reflexes have also been reported. [52] While majority of these patients are asymptomatic baring few exceptions. [53],[54] In Indian literature, the only publication has included various electrocardiographic changes in WD. Fifteen among the 50 patients were noted to have an abnormality and the ECG included sinus tachycardia, sinus bradycardia, bifid P wave, ST elevation, ST depression, T wave inversion, ventricular premature contraction and prominent U waves. [55] The authors concluded that these abnormalities were probably related to copper deposition in the myocardium. Imaging Imaging studies in WD have been carried over years and included X-rays, CT brain, MR imaging and spectroscopy. [56],[57],[58],[59],[60],[61],[62],[63] Roentgenograms The various skeletal abnormalities in our study included osteoporosis (38.8%); fractures (3.4%); arthritis (4.1%) and milkman pseudofractures (8.2%). In our study, renal stones or gallstones were detected by ultrasonography and/or X-rays in 4.9 and 2.8% patients, respectively. [1] Computed tomography (CT brain) Prior to widespread availability of MRI facilities, CT scan was the imaging modality of the brain. However CT grossly underestimates the pathology of WD. In a series of 116 patients of WD, the observed CT abnormalities included: Cortical atrophy (44.8%), ventricular dilatation (44%), caudate atrophy (25%), brain stem atrophy (31.9%), cerebellar atrophy (19%) and hemispheric (29.3%), basal ganglionic (19.8%), thalamic (10.3%) hypodensities. [1],[56] Magnetic resonance imaging of brain Introduction of MR imaging has greatly helped the clinicians to understand the pathological and anatomical correlates of clinical manifestations in WD. MRI of the brain can assist in the diagnosis and may also help in prognostication. In a large study of MRI in 100 patients with WD, the salient findings included: Atrophy of the cerebrum (70%), brainstem (66%) and cerebellum (52%), signal abnormalities in putamen (72%), caudate (61%), thalami (58%), midbrain (49%), pons (20%), cerebral white matter (25%), cortex (9%), medulla (12%) and cerebellum (10%). The characteristic 'face of giant panda' sign was noted in 12% and feature of central pontine myelinolysis was noted in 7% and bright claustral sign in 4% of patients. [57] From the same center, sequential MRI changes during the course of the treatment were studied in 50 patients. In this study the imaging features had improved in 70% of patients, not changed in 20%, worsened in 8% and the changes were both improvement and worsening in 2% of patients, improvement in imaging features, 20%; had no significant changes, 8%; had worsening of imaging features in 2%. These observations suggest that MRI can be used as a tool to monitor the therapy in WD. [58] The important MRI features that distinguish WD from other movement disorders are the presence of central pontine myelinolysis-like changes and midbrain tectal plate signal changes. [59],[64] [Figure - 3],[Figure - 4],[Figure - 5],[Figure - 6] Magnetic resonance spectroscopy provides insight into the biochemical changes in WD. Study of MRS of basal ganglia showed reduced breakdown and/or increased synthesis of membrane phospholipids and increased neuronal damage in patients with WD. [65] However, the exact clinical significance of these changes is yet to be established [Figure - 4]b. Disease Severity Assessment Scales As the WD has a dynamic course and variable response to treatment, disease severity assessment scales are required to monitor the disease course during longitudinal follow-up studies. Several assessment scales and scores have been used in WD and these include: Neurological Symptom Score and Chu Staging to stage the disease and assess the clinical outcomes; Hepatic Prognostication Index for assessing biochemical prognostication; MRI Scoring Scale for assessing the imaging prognostications and WHO Quality of Life scales among others. Recently, Aggarwal et al., from Mumbai have devised a new novel scale called the Global Assessment Scale (GAS) for the global assessment of WD. It is yet to be validated in a larger cohort of patients. [66] Therapeutic Aspects The detail descriptions of the disease by Wilson in 1912, did not offer any therapeutic options due to lack of understanding of the pathophysiology. The diagnosis of WD was considered as a road to an inevitable death within 5-10 years depending on the clinical presentation. The outcome remained gloomy until 1948 when Cuming described deposition of copper in various body organs and suggested usage of Dimercaprol (BAL) for chelating the copper. This breakthrough in management was followed by introduction of penicillamine as de-coppering therapy in 1955 by Walshe. A number of drugs such as zinc, trientine and ammonium tetrathiomolybdate have been shown to have beneficial effect and have been frequently used in the management. Liver transplantation, the most definitive treatment for WD has been successfully carried out in 1982. A general approach to patients with WD is provided in [Figure - 7]. Indian experience of management of WD is varied and has involved use of dimercaprol, penicillamine, potassium sulfide, and zinc. [1],[5],[6],[67],[68],[69] Surgical interventions have also been tried. Dimercaprol (BAL) Dimercaprol was the first drug to be introduced in the treatment of WD in India. However, Dastur et al., (1968) did not find any significant improvement with the treatment in six of their patients. Of the six patients only three received the drug for more than 3 months. [6] In another study by Singh et al., (1978), from Pondicherry all the 8 patients treated with BAL were symptomatically better. [67] However, following introduction of penicillamine, the use of BAL has virtually become exceptional. Penicillamine Penicillamine is the most commonly used de-coppering drug in India. Use of penicillamine has been reported to cause significant and paradoxical worsening during the initial phase by a few authors, [70] but there are no systemic studies to support this view in the Indian literature. In our experience, only a few cases exhibit paradoxical worsening. This can be significantly reduced if penicillamine is started in a low dose and built up to the required dosage slowly. Some of the rare adverse events like Steven Johnson syndrome, [68] myasthenia gravis [71] and pseudoxanthoma elasticum [72] have also been documented. To ascertain penicillamine-induced impaired neuromuscular transmission, a prospective study involving 60 patients of WD was carried over at NIMHANS. The parameters included were bedside fatigability tests, repetitive nerve stimulation tests and serum AChR antibodies titers in drug naοve as well as patients receiving penicillamine. It was concluded that long-term use of penicillamine did not cause any clinical, electrophysiological or immunological evidence of myasthenia in patients with WD. [73] In our series of 16 patients with 30 successful pregnancies, teratogenicity was not reported with low dose penicillamine and zinc. [69] Zinc Zinc was introduced in the management of WD in the early 1960s. Since then a significant number of studies on its usage have been published. Zinc has no or minimal side effects and therefore forms an essential component of treatment in vast majority of patients. Zinc therapy in India is gaining significant value because of economical reasons and encouraging results in the recent literature. Unlike penicillamine and other de-copppering agents, zinc is cheap, easily available and therefore patients have better compliance. In the first study in India of zinc therapy by Murthy et al., of the eight patients started on maintenance zinc therapy, seven patients had sustained improvement. [68] In another study by Sinha et al., 45 patients were initially started on both penicillamine and zinc sulfate, but subsequently had to be shifted to zinc as the lone agent because of economic reasons and were followed for the next two-and-half years. None of them had any significant deterioration in the clinical status while on zinc therapy alone. The authors concluded that zinc can be used as an effective maintenance therapy in Indian population. [69] However, in a recent study clinical and biochemical progression was observed in two asymptomatic siblings who were started on zinc as a lone therapy. Both these patients improved clinically and biochemically following institution of penicillamine. [74] Thus there is need to have trials of zinc alone as initial therapy in symptomatic patients in Indian population. Trientine and ammonium tetrathiomolybdate The experience in Indian literature with trientine dihydrochloride, which has been in use for more than three decades all over the world, is very limited. This is mainly because of the high cost and nonavailability of the drug. Further, relative safety and efficacy of the combination therapy of penicillamine and zinc has also limited its usage. However, trientine has been used in a few cases wherein penicillamine was not tolerated or has been found to have paradoxical worsening. [1] To our knowledge, reports of ammonium tetrathiomolybdate in WD have not been published from India. Surgical modalities Surgical intervention has been limited in WD as most of the symptoms are potentially reversible with medical therapy. However, certain symptoms like disabling tremors are very much resistant to drug therapy and may require surgical intervention. Only a few case reports of surgery in WD have been reported in the literature. [6],[27],[75] The most common surgery was thalamotomy for resistant tremors. [76] Special Issues There are certain gender (female) related issues in the management of WD. In our large series, of the 59 pregnancies in 16 patients with WD, 30 pregnancies were successful, 24 pregnancies resulted in spontaneous abortion, two medical terminations and three still births. Of the 16 women, 10 women were pre-symptomatic and the diagnosis of WD was established after conception and the remaining six were on treatment for WD. Among these 16 patients, 9 had history of spontaneous abortions and 12 had successful pregnancies. In none of the women there was a change in the clinical features during pregnancy with or without therapy. Our results suggest that pregnancy does not have adverse effect on the course of WD. In this series, we have not observed any teratogenic effects in the babies born to mothers with WD on de-coppering therapy. [77] Recent studies have documented sleep disorders in patients with WD. A study using sleep questionnaires and polysomnography (PSG) in patients with WD suggested high frequency of sleep disorders on the sleep questionnaire. On PSG study, patients had significantly reduced total sleep-time, decreased sleep-efficiency, increased sleep-onset latency and latency to stage-2 and reduced percentage of deep-sleep and REM-sleep. [78] Outcome Measures The best outcome measure of any therapeutic intervention in any disease is the changes in the quality of life (QoL). This is particularly so in chronic diseases like WD. The outcomes following specific therapeutic intervention can broadly be divided into: (a) total cure with normal QoL following therapeutic intervention or disease stabilization with some issues in QoL; (b) relentlessly progressive course of the disease in spite of specific therapeutic intervention with poor QoL and (c) initial stable course of the disease following specific therapeutic intervention and then dramatic worsening with deteriorating QoL. Most of the patients with WD lead normal life following the institution of specific medications. [79] In the study by Prashanth et al., of the 140 patients with WD on regular medication and follow-up, 126 (90%) patients had good clinical outcome. In this study some of the patients with severe illness at the start of medication had good response to the de-coppering therapy and 50% of patients with severe illness improved variably with de-coppering therapy. It was difficult to differentiate between the poor and good responders at the initial evaluation. [80] However, it has been observed that patients with white matter lesions on MRI may have poor response to de- coppering therapy [80],[81] . Rapid progressive course is usually noted in patients with hepatic form of the disease e.g. acute fulminant hepatic failure. A subset of patients with unpredictable course certainly pose management problem. Some patients may have very stable initial course with dramatic response to de-coppering agents and may suddenly deteriorate during the clinical course of the illness, usually due to poor drug compliance and sometimes by inter-current infections and stress. The response to therapy is rather poor in patients who have deteriorating course on treatment. The exact reason for this delayed worsening remains a challenge. These patients probably have very delicately maintained internal milieu and any factor that tends to change the balance leads to vicious cycle of deterioration. Conclusions WD is perhaps more common than reported from India. There is a need for epidemiological studies and also multicentric genetic study in view of high degree of consanguinity in certain parts of India, particularly in south India. A nationwide registry may improve awareness among medical communities and lead to early diagnosis and prompt treatment. Once diagnosis is established the patient and the caregiver need to be educated regarding compliance and long-term follow- up. Governments, both central and state, should see that the drugs are made available at subsidized prices as WD is a potentially curable disease. Acknowledgments We are grateful to the staffs, residents and faculty of various departments at NIMHANS, Bangalore for their contributions, assistance and involvement in the care of patients and various studies spanning over four decades. Not the least, we are very thankful to our patients who consented for the research work. References
Copyright 2009 - Neurology India The following images related to this document are available:Photo images[ni09161f5.jpg] [ni09161f7.jpg] [ni09161f4.jpg] [ni09161f3.jpg] [ni09161f1.jpg] [ni09161f2.jpg] [ni09161f6.jpg] [ni09161t1.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}