 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
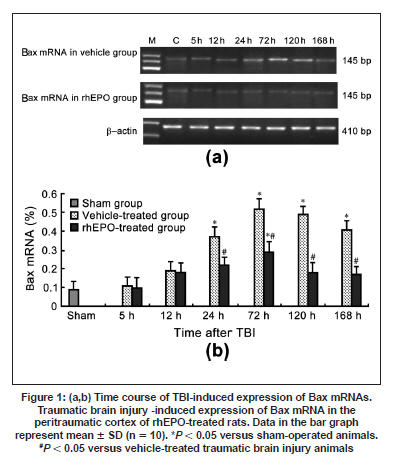
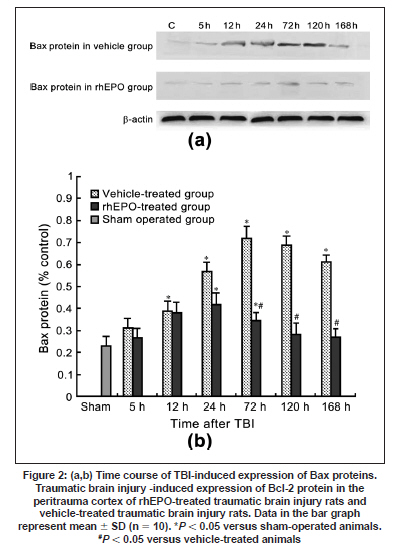
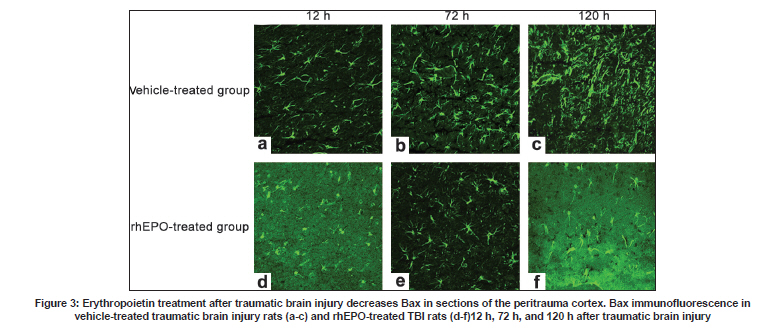
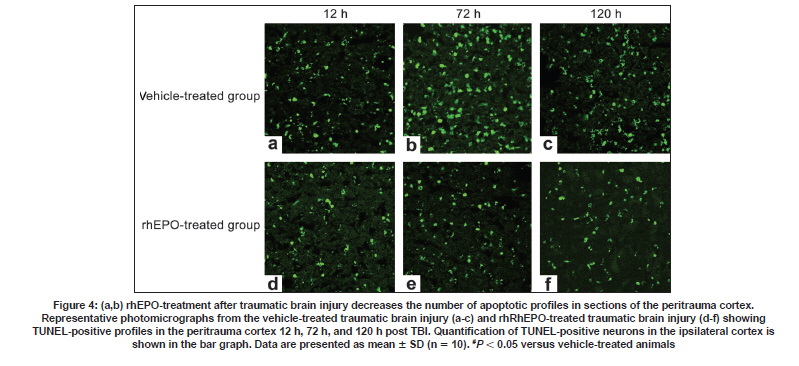
Neurology India, Vol. 57, No. 6, November-December, 2009, pp. 722-728 Original Article Erythropoietin can promote survival of cerebral cells by downregulating Bax gene after traumatic brain injury in rats Z. B. Liao, G. Y. Jiang, Z. H. Tang, X. G. Zhi, X. C. Sun, W. Y. Tang, M. J. Wu1 Departments of Neurosurgery, The First Affiliated Hospital of Chongqing Medical University, Date of Acceptance: 02-Jul-2009 Code Number: ni09206 PMID: 20139499 DOI: 10.4103/0028-3886.59466 Abstract Background : Traumatic brain injury (TBI) is an important cause of adult mortality and morbidity. Erythropoietin (Epo) has been shown to promote the viability of cerebral cells by upregulating Bcl-2 gene; however, Epo may exert its antiapoptotic effect via the differential regulation of the expression of genes involved in the apoptotic process.Aim : The present study examined the neuroprotective effect of Epo as a survival factor through the regulation of the Bax. Materials and Methods : Wistar rats were randomly divided into three groups: Recombinant human EPO treated (rhEPO) TBI, vehicle-treated TBI, and sham-operated. Traumatic brain injury was induced by the Feeney free-falling model. Rats were killed 5, 12, 24, 72, 120, or 168 h after TBI. Regulation of Bcl-2 was detected by reverse transcription-polymerase chain reaction (RT-PCR), western blotting and immunofluorescence. Results : Bax mRNA and protein levels were lower in the rhEPO)-treated rat brains than in the vehicle-treated rat brains. Induction of Bax expression peaked at 24 h and remained stable for 72-120 h in vehicle-treated rat brains, whereas induction of Bax expression was only slightly elevated in rhEPO-treated rat brains. The number of TdT-mediated dUTP Nick-End Labeling(TUNEL)-positive cells in the rhEPO-treated rat brains was far fewer than in the vehicle-treated rat brains. Conclusions : Epo exerts neuroprotective effect against traumatic brain injury via reducing Bax gene expression involved in inhibiting TBI-induced neuronal cell death. Keywords: Bax, cell death, rhEPO, traumatic brain injury Introduction The high incidence of adverse outcomes following traumatic brain injury (TBI) has been attributed to a large extent to the secondary mechanisms of neuronal cell death. [1],[2] These mechanisms include induction of neuronal cell death and complement-mediated neuronal cell lysis. [3],[4],[5],[6],[7] A number of studies have demonstrated that TBI-induced neuronal cell death is crucial for secondary injury, and anti-apoptotic effects may account in large part for the protective effects. [8],[9] Neuronal and glial cell death contribute to the overall pathology of TBI in both humans and animals. In both experimental head-injury models and human TBI, apoptotic cells and degenerating cells exhibiting classic necrotic morphology have been observed. [10],[11] Neuroprotective strategies for prevention of the neuropathological sequelae of TBI have largely failed in translation to clinical treatment. [12],[13],[14] Thus, there is a need for further understanding of the molecular mechanisms of the drugs which inhibit neuronal cell death in the injured brain. [15] Erythropoietin (EPO) is a cytokine that was originally identified as the major regulator of erythroid precursor cells. [16],[17],[18] However, increasing evidence suggests that EPO has broader functions independent of its effects on erythropoiesis. EPO has a neuroprotective effect in animal models of global and focal cerebral ischemia, as well as in adult rodent models of spinal ischemia, [19] Although the exact mechanism of neuroprotection is unknown, EPO likely promotes signaling cascades involved in cell survival and/or upregulation of antiapoptotic proteins. [20] In our earlier study we have shown enhanced erythropoietin- receptor expression for almost a week after TBI, where as EPO was slightly elevated, that too in the first 72 h only. [21] EPO administration protects neurons by enhancing Bcl-2 expression and thus inhibits TBI-induced neuronal cell death. However, EPO may exert its antiapoptotic effect via the differential regulation of the expression of genes involved in the apoptotic process. In the present study we explored the regulation of Bax gene after TBI, as well as the anti-apoptotic effects of recombinant human EPO (rhEPO) treatment. Materials and Methods Animal experiments Wistar rats (180-210 g, Chongqing Medical University, China) were housed under 12-h periods of light and darkness with free access to food and water. All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80-23, revised 1996). The number of animals used and their suffering were minimized. TBI (1000 g-cm) was induced in anesthetized rats using the weight-drop contusion model as described. [9],[10] Briefly, 120 rats were anesthetized using sodium pentobarbital (60 mg/kg i.p.) and underwent a craniotomy, in which a circular region of the skull (3.0 mm diameter, cantered 2.3 mm caudal and 2.3 mm lateral to bregma) was removed over the right somatosensory cortex. A weight-drop device was placed stereotactically over the dura and adjusted to stop an impact transducer (foot plate) at a depth of 25 mm below the dura. Then, a 40-g weight was dropped from 25 cm above the dura, through a guide role, onto the foot plate. Body temperature was also monitored during surgery with a rectal probe and maintained in the range of 36.5-37.5°C with a heating pad. After the injury, the scalp was sutured. No trauma was induced in the sham-operated group. Rats were killed 5, 12, 24, 72, 120, and 168 h after TBI. Animal groups Rats were randomly divided into three groups: Recombinant human EPO (rhEPO) treated TBI (n = 60), vehicle-treated TBI (n = 60), and sham-operated (n = 10). EPO (5,000 IU/kg; 3cbio, Shengyang province, China) or vehicle (2 mL containing 2.50 mg human serum albumin, 5.84 mg sodium chloride, 1.164 mg sodium phosphate monobasic dehydrate, and 2.225 mg sodium phosphate dibasic dehydrate per mL) was administered intraperitoneally once a day for seven days after TBI. Doses of EPO were selected based on previous studies. [11] Tissue samples were collected from ten EPO- and ten vehicle-treated rats at 5, 12, 24, 72, 120, and 168 h after TBI. The sham-operated tissue samples were collected 168 h after TBI. Tissue processing Rats were killed by decapitation and the brains were rapidly dissected, frozen on liquid nitrogen, and stored at -80°C for biochemical analysis for T dT-mediated d U TP N ick- E nd L abeling (TUNEL) staining and immunofluorescence, Rats were deeply anesthetized with sodium pentobarbital (60 mg kg i.p.) and then transcardially perfused with 400 ml 4% paraformaldehyde and 100 ml 30% sucrose. Fixed brains were removed and stored at -80°C. RT-PCR Brain tissues from the peritrauma cortex (surround the impact point from 1 cm to 1.5 cm) in rhEPO-treated TBI, vehicle-treated TBI, and sham-operated rats were rigorously homogenized. Total RNA from the peritrauma cortex tissue was isolated using the TRIzol method. For Bax RT-PCR amplification, the forward and reverse primers were 5'-CTCAGAAGGAATTGATGTCG-3'and 5'-TCACGCCAACCCTCCACCAC-3'respectively, yielding a145-bp PCR product. β-actin was used as an internal standard; the forward and reverse primers were 5'-ATGCATCCTGCACCACCAAC-3'and 5'- TGGAGAAACCTGCCAAGTAT-3'respectively, yielding a 410-bp PCR product. The reaction was maintained at 42°C for 60 min, then at 92°C for 5 min before 35 cycles of: 94°C for 30 s, 53°C for 30 s, and 72°C for 60 s, followed by 72°C for 10 min. PCR products were electrophoresed on a 1.5% agarose gel and stained with ethidium bromide (2 µg/mL). Western blotting Brain tissues from the peritrauma cortex in rhEPO-treated TBI, vehicle-treated TBI, and sham- operated rats were rigorously homogenized and sonicated for 30 s in a lysis buffer as described. [6] Protein concentrations were determined and aliquots of 10 ug of the total proteins were separated by electrophoresis on SDS- polyacrylamide gels. Briefly, protein samples were separated by electrophoresis on an 8% polyacrylamide gel and electrotransferred to a nitrocellulose membrane. Non-specific binding sites were blocked by incubating overnight at 4°C in TBS containing 2% BSA and 0.1% Tween-20. Membranes were rinsed for 10 min in buffer (TBS containing 0.1% Tween-20) and then incubated with anti-Bax, anti-β-actin, (1:2000, Santa Cruz Biotechnology anti-rat) followed by anti-goat IgG horseradish peroxidase-conjugate (Santa Cruz Biotechnology). After rinsing with buffer, the immunocomplexes were visualized using an enhanced chemiluminescence kit (Amersham Pharmacia Biotech Inc., Piscataway, NJ) according to the manufacturer's instructions. Film signals were digitally scanned and then quantified using NIH Image software. β-actin was used as an internal control for all Western blots. Immunofluorescence The peritrauma coronal sections of 10-um thickness were cut with a cryostat and mounted on slides. Cryosections were fixed in 1% paraformaldehyde in PBS and postfixed in a mixture of ethanol and acetic acid (2:1) at 20°C. Sections were washed in PBS and blocked by incubating in PBS containing 0.3% Triton X-100 and 2% BSA. Bax were localized with rabbit anti-Bax (Santa Cruz Biotechnology anti-rat) antibody at 1:2000. Bound primary antibodies were detected by incubation with fluorescein isothiocyanate (FITC)-coupled anti-rabbit IgG at 1:2500. Controls were performed by omission of the primary antibody. And the results of immunofluorescence controls were negative as no staining was observed in peritrauma cortex tissue. Sections were observed using the Aristoplan confocal laser-scanning microscope. Neuronal cell death assay The peritrauma coronal sections of 10-um thickness were cut with a cryostat. Neuronal cell death in the peritrauma cortex was examined with a commercial TUNEL assay kit (Santa Cruz Biotechnology) that enzymatically labels DNA strand breaks. Slides were incubated with blocking solution for 10 min at 20°C to eliminate endogenous peroxidases, followed by permeabilization solution for 2 min on ice. Then the TUNEL reaction (50 µL) was added and the slides were incubated in a humidified atmosphere in the dark for 1 h at 37°C. As a negative control, sections of the peritrauma cortex were used after the standard procedures, but labeled dUTP was omitted. Results are expressed as counting T dT-mediated d U TP N ick- E nd L abeling (TUNEL) labels apoptotic cells. Sections were observed using the Aristoplan confocal laser-scanning microscope. TUNEL labels , apoptotic cells were counted at five different fields in the inner border of the peritrauma cortex in five sections per rat over a microscopic field of 0.01 mm 2 , and averaged and expressed as cells/0.01 mm 2 . Five rat/group were analyzed by an observer blind to the experimental treatment (see below). Image and data analysis After capturing images with a digital camera, quantification of the results from Immunofluorescence, Western blotting and RT-PCR was performed with National Institutes of Health IMAGE 1.61 software. The total number of TUNEL-positive cells per image (cells/0.01 mm 2 , objective x40) was calculated by an observer blind to the experimental treatment. In each section, five peritrauma cortical areas outside labeled neurons were chosen randomly to obtain an average value for the subtraction of background by an observer blind to the experimental treatment. Statistical analysis was performed by ANOVA. All results were expressed as mean ± SD. A P value of < 0.05 was considered to be statistically significant. Results Erythropoietin administration decreases Bax mRNA after traumatic brain injury Basal levels of Bax mRNA were observed in sham-operated brain tissue [Figure - 1]a (0.09 ± 0.021). In the vehicle- treated group, Bax mRNA was slightly upregulated at 5 h after TBI [0.11 ± 0.046;1.2-fold increase vs. sham, n = 5, P > 0.05; [Figure - 1]b], significantly upregulated at 12 h [0.19 ± 0.048; n = 5, P < 0.05 vs. sham; [Figure - 1]b], peaked at 24 h [0.37 ± 0.052, n = 5, P < 0.05 vs. sham; [Figure - 1]b] and persisted at peak levels up to 168 h post-TBI. We also evaluated the effects of EPO administration on Bax mRNA level in TBI rats. In the rhEPO-treated group, Bax mRNA expression in the ipsilateral peritrauma cortex peaked at 72 h [0.29 ± 0.053, n = 5, P > 0.05 vs. sham; [Figure - 1]b] and returned to near baseline by 120 h post-TBI [0.18 ± 0.056, n = 5, P > 0.05 vs. sham, [Figure - 1]b]. Erythropoietin administration decreases the expression of Bax protein after traumatic brain injury Next we determined whether EPO administration regulates TBI-induced Bax protein expression. Sham-operated samples expressed basal levels of Bax (0.23 ± 0.043). Relative to the sham group, Bax in the vehicle-treated TBI was elevated 1.3-fold [0.31 ± 0.046, n = 5, P > 0.05; [Figure - 2]]]] at 5 h after TBI, reached a peak at 72 h [0.72 ± 0.051;3.1-fold, n = 5, P < 0.05; [Figure - 2]]], and was sustained to 168 h [0.61 ± 0.046;2.6-fold, n = 5, P < 0.05; [Figure - 2]]. In the rhEPO-treated TBI group, Bax was increased by 1.1fold [0.27 ± 0.051; vs. sham, n = 5, P > 0.05; [Figure - 2]] at 5 h post-TBI, 1.4-fold [0.34 ± 0.053, n = 5, P < 0.05; [Figure - 2]] at 72 h, and 1.2-fold [0.27 ± 0.042, n = 5, P > 0.05; [Figure - 2]] at 168 h. To determine the relative number of cells expressing Bax within the peritrauma cortex, immunofluorescence imaging was used. Bax was detected in many cells in the peritrauma cortex, many of which had morphological features characteristic of neurons. Strong Bax induction was observed throughout the peritrauma cortex from 24 h to 168 h post-TBI in the vehicle-treated group, whereas Bax expression was only observed transiently in the rhEPO-treated TBI group [Figure - 3], consistent with Western blot analysis. Weak Bax immunoreactivity was also detected in the superficial cortex of sham-operated rats (data not shown). Erythropoietin administration decreased the number of TdT-mediated dUTP Nick-End Labeling -positive neurons after traumatic brain injury To evaluate the potential anti-apoptotic effects of EPO, we measured the number of apoptotic profiles by TUNEL staining. No TUNEL-positive cells were observed in the sham. TUNEL-positive cells were noted in the vehicle-treated TBI, predominantly in the peritrauma cortex [Figure - 4]. The number of TUNEL-positive cells in the vehicle-treated TBI increased at 24 h [42 ± 5.59, n = 5, P < 0.05; [Figure - 4]] post-TBI and peaked at 72 h [68 ± 7.51, n = 5, P < 0.05; [Figure - 4]]. However, only scattered TUNEL-positive cells were observed in the rhEPO-treated TBI at 72 h [32 ± 5.48 n = 5, P < 0.05 vs. control; [Figure - 4]]. Cell death was relatively constant over the time course in the rhEPO-treated TBI. In some TUNEL-positive cells, a condensed, clumped, or pyknotic nuclear pattern was observed. Discussion Based on the morphological criteria and the TUNEL method, cerebral cell loss was characterized by both cell necrosis and cell death These observations are in agreement with the previous studies. TBI initiates a complex cascade of events that eventually result in injury and subsequent necrosis and/or apoptotic death of cerebral cells. [22] We demonstrated that EPO inhibits apoptotic cell death in peritrauma cortex as determined by DNA fragmentation, confirming previous data which suggested that inhibition of cell death is one of the most potential protective mechanisms of EPO. [23],[24],[25] In rats subjected to TBI, systemic administration of EPO for seven days post trauma significantly reduced the number of TUNEL-positive cells in the peritrauma cortex compared to the vehicle-treated group, suggesting that EPO affords protection by inhibiting cell death. Furthermore, in our earlier study, [21] our findings demonstrated that Bcl-2 protein is induced in peritrauma cortical cerebral cells in rhEPO-treated rats for 5 h post trauma and this effect was maintained up to 168 h, whereas in vehicle-treated rats the effect was maintained only for 72 h. Previous studies conducted in similar animal models of TBI have also shown that severe TBI leads to an increase in the Bax/Bcl-2 ratio suggesting that the fine balance between the activity of pro-apoptotic and anti-apoptotic Bcl-2 family members can determine cell survival and modulate the induction of cell death. [22],[23],[24],[25] In order to further clarify the anti-apoptotic mechanisms of EPO, we have also investigated the effect of TBI on the activation of Bax in the brain tissue with and without treatment of EPO. Bax is a cell-death gene implicated in the regulation of diverse biological phenomena, including cell death, cell survival and growth, cellular differentiation, hypoxia and ischemia. [26],[27],[28],[29],[30] The results in our study presented here demonstrates that TBI leads to activation of Bax, which suggests that it might be a marker of injury linked to the pathophysiology of TBI. [31] Induction of Bax expression peaked at 24 h and remained stable for 72-120 h in vehicle-treated TBI, whereas it was only slightly elevated in rhEPO-treated TBI. Roughly half of the cerebral cells in the peritrauma cortex of vehicle- treated TBI rats were Bax immunoreactive. Cerebral cells expressing Bax protein appeared to be peritrauma cortical neurons and glial cells. Administration of EPO also decreased the number of TUNEL-positive cells, suggesting that Bax plays an important role in regulating neuronal death and a complementary role as an inducible neuroprotective protein after TBI. i0 n vitro studies suggest that Bax possesses a key role in the phosphatidyl-inositol 3-kinase-Akt (PI3K-Akt) pathway that acts as a cell death signal in the regulation of cell death in neuronal or glial cells. [32],[33] It is already known from the recent literature that Bax plays a key role in the EPO-mediated cell death action in the nervous system. [34],[35],[36] It appears, therefore, that activation of Bax may serve a dual role in inhibiting or promoting cell death pathways in a cell type- and stimulus-dependent manner. [37] Based on the information in the literature, recently, data provided by in vitro studies suggests that EPO-mediated protection against cell death may involve cross-talk between JAK2 [38] and NF-kB resulting in an early (within 24 h of TBI) activation of NF-kB signaling pathways. [39] However, an EPO-triggering activation through a specific signal transduction pathway and regulatory mechanisms, such as types of activated Bax homodimers could ensure the transient activation of the cell death factor and subsequently promote its beneficial role. [40],[41],[42],[43] This might be one of the potential mechanisms underlying the protective action of EPO, which functions independently or in combination with other intracellular signaling cascades. Alternative protective mechanisms that might be activated downstream from the EPO/EPOR system could include the regulation of mitogen-activated protein kinase (MAPK) and the (PI3K/Akt) system, as well as the increased expression of HSP70. [44],[45],[46] Evidence to support these hypotheses is derived from observations of in vitro and in vivo studies of TBI- induced injury in the central nervous system(CNS.) [47] Our data demonstrated that EPO administration offered significant protection against TBI via reducing Bax gene and upregulating Bcl-2 gene [48] expression involved in inhibiting TBI-induced neuronal cell death. However, the mechanism by which EPO protects peritrauma cortical cerebral cells against TBI is not fully elucidated. Improved understanding of EPO-mediated signaling cascade is needed in order to delineate the benefits of EPO therapy and incorporate its potential use into clinical practice in the future. Based on these findings, EPO treatment may represent a novel therapeutic approach due to its capacity to preserve CNS function and directly protect peritrauma cortex from traumatic brain injury in rats. Acknowledgments This research was supported by the National Natural Science Funds of China (30801182). References
Copyright 2009 - Neurology India The following images related to this document are available:Photo images[ni09206f1.jpg] [ni09206f4.jpg] [ni09206f3.jpg] [ni09206f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}