 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
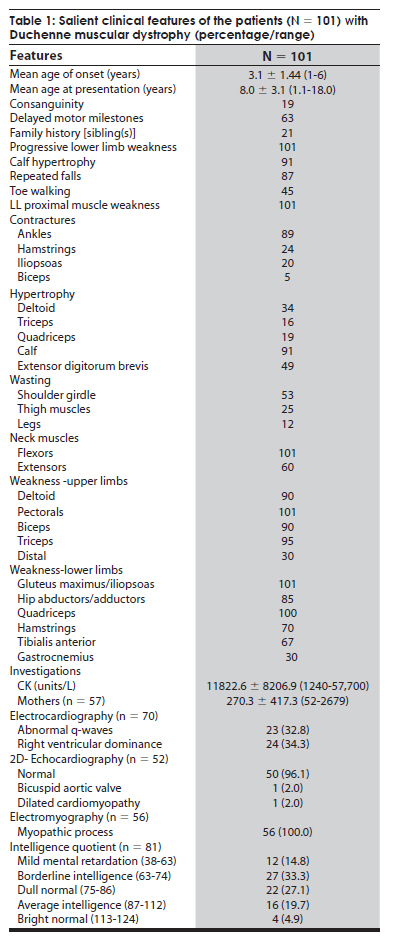
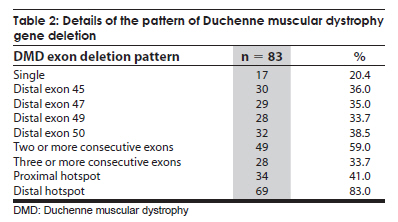
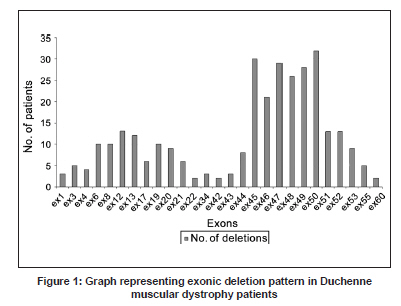
Neurology India, Vol. 57, No. 6, November-December, 2009, pp. 734-738 Original Article Duchenne muscular dystrophy: A clinical, histopathological and genetic study at a neurology tertiary care center in southern India Bhairavi Swaminathan, G. N. Shubha, D. Shubha, A. Ram Murthy, H. B. Kiran Kumar, S. Shylashree1 , N. Gayathri 2 , R. Jamuna3 , Sanjeev Jain, Meera Purushottam, A. Nalini 1 Departments of Molecular Genetics Laboratory, 1 Neurology, 2 Neuropathology, and 3 Neuropsychology, National Institute of Mental Health and Neurosciences, Bangalore, India Date of Acceptance: 25-Apr-2009 Code Number: ni09208 PMID: 20139501 DOI: 10.4103/0028-3886.59468 Abstract Background : Duchenne muscular dystrophy (DMD) is the most common muscular dystrophy that affects young boys and the dystrophin gene on the X chromosome has been found to be associated with the disorder.Materials and Methods : In this prospective study, 112 clinically diagnosed DMD patients had muscle biopsy and were tested for exon deletions. Genotyping was also carried out at STR44, STR45, STR49 and STR 50 markers in 15 families. Results : Of the 112 clinically suspected DMD patients, the diagnosis of DMD was confirmed by histopathology and/or genetics in 101 patients. The mean age of onset was 3.1±1.44 years (1-6 years) and the mean age at presentation was 8.0±3.1 years (1.1-18.0 years). Delayed motor milestones were present in 63 (62.3%) patients. The mean creatine kinase value was 11822.64±8206.90 U/L (1240-57,700). Eighty-four patients had muscle biopsy and immunohistochemistry was done in 60 muscle samples, all of which demonstrated absence of dystrophin staining. Of the 60 dystrophin-negative cases, 73% showed deletion of at least one exon. Single exon deletion was found in 20.4%. Distal hotspot Exons 45, 47, 49 and 50 were the commonly deleted xenons and the deletion rates were 36%, 35%, 33.7% and 38.5% respectively. Conclusions : In this study population in south India the deletion rate was 73% and were more frequent in the distal end exon. With the availability of genetic analysis, the first investigation of choice in DMD should be genetic studies and muscle biopsy should be considered only if the genetic tests are negative or not available. Keywords: Duchenne muscular dystrophy, dystrophin gene, exon deletions Introduction Duchenne muscular dystrophy (DMD) is the most common form of all muscular dystrophies with an incidence rate of 1:3500 live male births. [1] Earlier, histopathology was the most widely accepted method of distinguishing the types of muscular dystrophies. [1] Availability of genetic tests make it possible to diagnose these disorders early and also avoid invasive procedures like muscle biopsy. The gene responsible for DMD is one of the longest genes, spanning a length of 2.3 MB and contains 79 exons. [2] The disease is caused by mutations in the dystrophin gene, which leads to the loss of function of the protein. [3] Deletions account for 60-65% cases in DMD; duplications for 5-6% and point mutations for the remaining cases. [4],[5],[6],[7] Using primers targeting 18 hotspot exons in the dystrophin gene, 98% of deletions can be detected. The proximal hotspot encompasses Exons 3-7 and the distal hotspot Exons 45-51. [8] The genetic diagnosis for DMD involves multiplex polymerase chain reaction analysis of 27 exons which include these hotspots. Dinucleotide repeat polymorphism-based genetic analysis at short tandem repeats loci within the gene and near the deleted exon can ascertain carrier status in the majority of female relatives. [4],[9] The present study reports the analysis of exonic deletions in 112 clinically suspected DMD patients. Materials and Methods The study was approved by the Institutional Ethics Committee. A prospective study was performed on 112 definite or probable DMD patients. Diagnosis was based on clinical characteristics, elevated serum creatine kinase (CK) and electromyographic features. Other evidences for the diagnosis of DMD prior to genetic analysis included: The absence of dystrophin staining in muscle biopsy and X-linked inheritance pattern of a myopathy clinically compatible with DMD. General intelligence was tested using Binet Kamath Test of Intelligence. [10] Based on the intelligence quotient they were classified as mild mental retardation(38-63), borderline intelligence (63-74), dull normal (75-86), average intelligence (87-112), and bright normal (113-124). Muscle biopsy Muscle biopsies were obtained by the open method from quadriceps or biceps muscles. Immunostaining with monoclonal antibodies to dystrophin (1, 2, 3) (Novocastra, UK)., a-Sarcoglycan and a-2 Laminin (Merosin) as primary and HRP tagged LSAB as secondary was carried out. Genetic analysis After written informed consent, genomic DNA was isolated from blood by the salting out method as described by Miller et al. [11] Multiplex polymerase chain reactions (PCR) were carried out for 27 exons according to Chamberlain et al., and Beggs et al. [8],[12] PCR products were resolved on 9% PAGE gels or 2% Agarose gels, and the gels were analyzed for exonic deletions by the presence or absence of a corresponding band. Microsatellite analysis Genotyping was carried out at STR 45, STR 49 and STR 50, highly polymorphic dinucleotide (CA)n loci in the dystrophin gene. [9] The FAM-labeled products were electrophoresed on a 5% ABI377 sequencing gel. Results Clinical findings Of the 112 clinically suspected DMD boys, the diagnosis of DMD was confirmed by histopathology and immunohistochemistry and/or genetic studies in 101 boys. The clinical details are described in [Table - 1]. All boys presented with progressive proximal muscle weakness particularly of the lower limbs and the majority (90%) complained of calf muscle hypertrophy. Investigations Details of the investigation findings are given in [Table - 1]. The mean CK value in 57 mothers was 270.3 ± 417.3 U/L (52-2679) and in 20 mother the values were above the upper limit of normal (170U/L) with four individuals having more than 1000 U/L. Of the 56 sisters of the probands tested for serum CK, two had values of 7420 and 23,490 U/L and in the remaining the values were normal. Intelligence Quotient (IQ) done in 81 patients showed varying degrees of mental subnormality. Of the 101 patients for whom the genetic data was available, 84 (83.2%) patients had muscle biopsy. In 60 (71.4%) of these patients immunohistochemistry was done, all of them demonstrated absence of dystrophin staining. Among the 60 dystrophin-negative cases, 44 (73%) patients had deletion of at least one exon. Genetic findings The deletion pattern is given in [Table - 2]. Distal hotspot Exons 45, 47, 49 and 50 were common deletions [Figure - 1]. Correlation of deletions with immunohistochemistry results is depicted in [Figure - 2]. Mild mental retardation was seen 15% of patients and all of them had at one deletion; 32.1% had borderline intelligence and 80.8% of them had at least one deletion; 27.2% had dull normal IQ and 81.8% of them had deletions; 19.8% had average intelligence and 75% of them had deletions, 5% were with bright normal IQ and 75% of them had deletions. Carrier testing Genotyping was undertaken at four short tandem repeat markers reported to have high heterozygosity viz. STR44, STR45, STR49, STR50 in the hotspot region of the dystrophin gene for 'mother and son' samples from 15 families. Six mothers were found to be carriers based on homozygosity at these marker loci. In these six families, positive family history was present in three and in the remaining three the deletions were probably new. Carrier status determination was used to study a DMD family where there was apparent paternal inheritance. [13] Discussion This study presents the clinical, histopathological, immunohistochemical and molecular findings in a large cohort of 101 patients with DMD from south India. Random distribution of age of onset was noted in the present study similar the earlier studies. [14] The distribution of motor weakness was similar to that described earlier. [15] Inter-individual variability in the clinical severity was observed among the patients and also with the families similar to the other studies. [16],[17],[18],[19] The reported frequency of the dystrophin gene deletions have been 22% to 86%. [6],[20],[21],[22],[23],[24],[25],[26] and dystrophin can be demonstrated by immunohistrochemistry in about 98% of DMD patients. [27] In our cohort the diagnosis of DMD was confirmed by either the absence of dystrophin staining only (18%) and/or exon deletions (82%). Correlation between exonic deletion and certain clinical features like ambulation, mental retardation, and histological findings have been observed Mental retardation is seen in about one-third of DMD patients. [23],[28] In our cohort mental retardation was seen in 48% of the patients, The lack of correlation between exonic deletions and immunohistochemical findings in 26% of the patients may be related to deletions in any of the other exons, mosaicism, or point mutation which we have not studied. Such genetic abnormalities have been documented in about 30% of cases. [4],[5],[6],[7] In most of the studies, 80% to 91% of deletions occurred in the distal region of the dystrophin gene [6],[24],[25] and the deletion rates can be low, 42% to 52%. [6],[20],[22],[24] In the study of 160 Indian patient population from all over the country, the deletion rate was 64.4% and 69.7% of which was in the distal hotspot region. This study did not find any ethnic differences in the deletion patterns of the dystrophin gene. [29] In the eastern Indian study the deletion rate was 63% and 79% of which was in the distal hotspot region. [30] Singh et al., reported a deletion rate of 73% in a north Indian patient population which included both DMD and Beckers muscular dystrophy. [25] Among a southern Indian DMD population the reported deletion rate was 62.1% and 78% of which was located in the distal hotspot region. [31] The authors of this study concluded that the lower deletion rate in their population when compared to the north Indian population may be related to the ethnic differences in the two populations. The deletion rate reported among 25 western Indian DMD patients was 72% mostly located at the 3' hotspot region. [32] The deletion rate in our study was about 73% and was similar to the frequency reported from the other parts of India. [33] The reported frequency of dystrophin gene deletions in other countries in Asia is quite variable: 40.7% in Pakistan, [34] and 66.25% in China. [35] In Egypt, an African country, it was 61.1%. [36] The diagnosis of DMD is based on clinical, biochemical and histopathologic studies and further confirmed by molecular analysis. However, genetic studies should be the investigations of choice in DMD and muscle biopsy should be limited to the cases where genetic studies are not informative. Carrier state assessment and prenatal diagnosis are essential for counseling and can be offered only after the possible mutation has been identified in the proband. Acknowledgment The authors acknowledge the Indian Council of Medical Research (ICMR), New Delhi for financial support extended to the 'Central Molecular Genetics Laboratory for Neurology and Psychiatry'. They also thank all the members and trainees in the laboratory who helped during the course of the work, especially Ms. Sukanya, Ms. Savitha, Ms. Sumita and Ms. Tanya. References
Copyright 2009 - Neurology India The following images related to this document are available:Photo images[ni09208f2.jpg] [ni09208t2.jpg] [ni09208t1.jpg] [ni09208f1.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}