 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
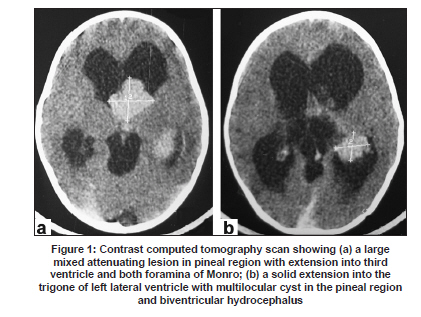
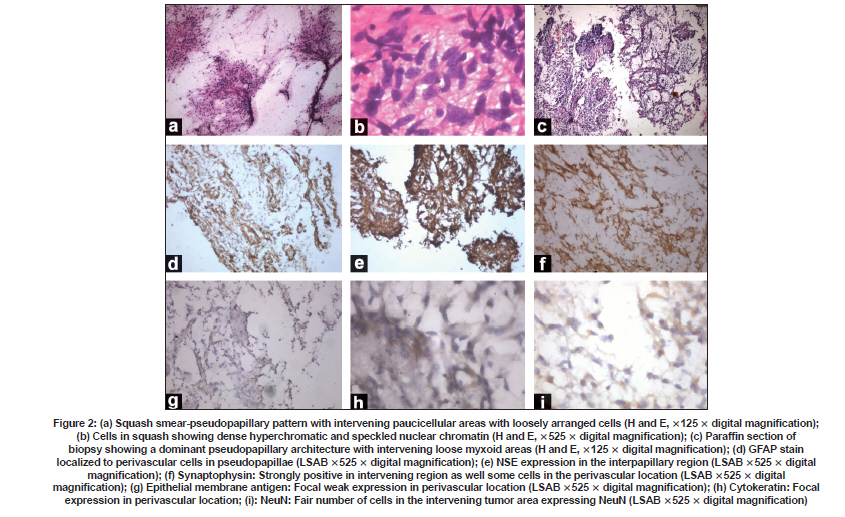
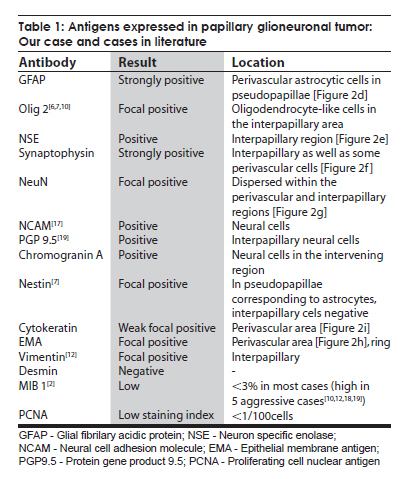
Neurology India, Vol. 57, No. 6, November-December, 2009, pp. 792-795 Case Report Endoscopic diagnosis of a pineal papillary glioneuronal tumor with extensive ventricular involvement: Case report with review of literature Nuzhat Husain, Mazhar Husain Department of Pathology and Neurosurgery, CSM (erstwhile King George’s) Medical University, Lucknow - 226 003, India Date of Acceptance: 27-Feb-2009 Code Number: ni09221 PMID: 20139514 DOI: 10.4103/0028-3886.59481 Abstract Papillary glioneuronal tumor (PGNT) is a newly described mixed glioneuronal tumor, recently included in the World Health Organization classification of central nervous system tumors. We report morphologic and immunohistochemical characteristics PGNT of the pineal region extending into the third and lateral ventricles in a four-year-old male child and reviewed all the cases reported in the international literature till date. In this child the diagnosis of PGNT was established by endoscopic biopsy and squash smear examination.Keywords: Endoscopic, glioma, glioneuronal, pineal, papillary Introduction Papillary glioneuronal tumor (PGNT) is a mixed glioneuronal tumor, recently included in the World Heath Organization (WHO) classification of central nervous system (CNS) tumors. Till date approximately 39 cases of PGNTs have been reported in the international literature. [1],[2],[3],[4],[5],[6],[7],[8],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18],[19] Credit for the first description of PGNT is given to Komori et al. who first described a series of nine cases in 1998. [2] The tumor has been assigned a WHO grade I, however aggressive variants have been reported. [9],[18],[19] We report morphological and immunohistochemical characteristics of PGNT in ventricular location diagnosed by endoscopic biopsy and squash smear. Case Report A four-year-old male child was admitted with symptoms of severe headache and projectile vomiting of ten days duration. Neurological examination showed bilateral papilledema and otherwise neurological and physical examination was unremarkable. Hematological and biochemical parameters were within normal limits. Contrast computed tomography (CT) scan showed a large enhancing mass lesion with cystic components in the pineal region with extension into third ventricle and both foramina of Monro and also into the trigone of left lateral ventricle with obstructive hydrocephalus [Figure - 1]a and b. The patient was subjected to endoscopic biopsy of the tumor. At the same sitting he had septostomy and a ventriculoperitoneal shunt for the relief of hydrocephalus. Postoperatively, the patient had uneventful recovery. He was relieved of physical symptoms. The patient's guardians refused resection surgery, No radiotherapy or chemotherapy was initiated and he was discharged with an advice to come for regular follow up. Histological studies Endoscopic biopsy from the tumor was processed in alcohol-fixed squash smears as well as paraffin-embedded formalin-fixed sections. The tumor irrigation fluid was collected along with the ventricular cerebrospinal fluid (CSF). Cytology smears were made from centrifuged fluid and stained with Giemsa and H and E stains. Paraffin sections from tumor biopsy were stained with H and E and immunohistochemical detection of antigen expression including glial fibrilary acidic protein (GFAP), neuron specific enolase (NSE), synaptophysin, epithelial membrane antigen (EMA), cytokeratin, NeuN, and proliferating cell nuclear antigen (PCNA) was done. All antibodies were obtained from Dakopatts, Denmark, except NeuN which was purchased from Chemicon International, USA. Detection was done with secondary LSAB 2 Kit (Dakopatts, Denmark), using DAB as substrate. H and E stained squash smears showed a papillary tumor with perivascular rosettes and intervening loose myxoid tissue [Figure - 2]a. Alcian blue and periodic acid Schiff's stains were negative for presence of perivascular mucin. The cells showed moderate degree of nuclear pleomorphism with perivascular cells showing dense hyperchromatic nuclei while cells at the periphery had speckled chromatin [Figure - 2]b and c. Smears of postoperative CSF and irrigation fluid also showed clusters of similar ells. Histological examination of the tissue revealed pseudopapillary structures with central hyalinized vessels and perivascular cell palisades positive for GFAP expression. Intervening regions showed sheets of loosely arranged cells. These cells showed round nuclei with speckled chromatin and were diffusely positive in immunohistochemistry for synaptophysin and NSE stains. Mature neuronal elements were not evident. The tumor displayed moderate cellularity and was devoid of necrosis, microvascular proliferation, mitoses, and calcification. Both cytokeratin and EMA showed focal weak expression in perivascular location. NeuN, a marker for primitive neuronal cells, was expressed in fair number of cells in the interpapillary zone. The tumor proliferation index was low with < 1% expression of PCNA. Discussion PGNT is relatively a rare tumor and occurs predominantly in young adults (mean age 27.2 + 18.36) with equal gender distribution. [1],[2],[3],[4],[5],[6],[7],[8],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18],[19] Three case including our case were aged less than five years of age, all three were aged four years at the time of presentation. [12],[13] The tumor distribution and extension is mostly cerebral: Temporal (13), frontal (12), fronto-pariental (1), parietal (6), parietoccipital (6), temporoparietal (2), and occipital (1). Ventricular location was in two patients including our patient. Our patient had solid and cystic component in the pineal region with extension to the third and lateral ventricles. Clinical presentation is quite variable and depends on the size and location of the lesion, headache being the common symptom. Seizures are more frequent with temporal lesions. [1],[2],[3],[4],[5],[6],[7],[8],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18],[19] Neuroimaging, both MRI and CT, features of the tumor form differential diagnosis of a cystic lesion with a mural nodule. Size of the lesion is highly variable, 1.0 cm [4] to 9 cm [19] with mean size of 4.7 cm. [1],[2],[3],[4],[5],[6],[7],[8],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18],[19] The lesions in most of the reported cases were partly cystic-enhancing masses. [1],[2],[3],[4],[5],[6],[7],[8],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18],[19] Of the 40 reported cases including our case, two cases had tumor-related hemorrhage. [6],[9] Lesions were frequently heterogeneous in non-contrast CT scan with cyst enhancement in the contrast CT. In MRI scan the lesions were hypointense on T1-weighted images, iso- or hyperintense on T2-weighted images and most tumors show heterogeneous enhancement in gadolinium. [3],[8] Diagnostic histological features include a pseudopapillary lesion with distribution of glial and epithelial markers in the pseudopapillary area with central hyalinized vessels. Intervening region with loosely dispersed sheets of cells shows strong expression of neuronal markers. These hallmarks of the lesion could be well distinguished in the small fragmented endoscopic biopsy in our case. In squash smears the lesion strongly resembled a myxopapillary ependymoma. Additional histological features which may be observed are presence of oligodendroglial cells in the pseudopapillary lining, [6] oligodendroglial-like cells within the interpapillary regions, as well as calcification. [6],[18] Rosenthal fibers, hemosiderin-laden macrophage, lymphocytic infiltrate have been described in the surrounding reactive tissue. [3] Additionally, presence of angiomatous thin-walled vessels in closely placed clusters has also been reported. [15] Surrounding tissue was not present in our case since the diagnosis was established in an intraventricular endoscopic biopsy. In addition to NSE and synaptophysin we have also observed presence of NeuN expression. NeuN positive cells comprised 44% of tumor cells in our case. NeuN positivity has also been observed in two previously reported cases of PGNT. [7] In a comparative analysis of NeuN expression in all major primary brain tumors, Preusser et al., have observed that NeuN expression may be helpful in the differential diagnosis of clear cell primary brain tumors but does not seem to be useful for the differential diagnosis of other brain tumor subtypes where it is variably expressed. [20] Since in the developing brain neuronal differentiation is associated with permanent exit from the mitotic cycle, Englund et al., postulated that neuronal differentiation may suppress proliferative activity even in neoplastic cells. They have observed that neuronal differentiation is associated with a substantial decrease of proliferative activity in neoplastic cells of central neurocytomas, and suggest that the same may be true across diverse types of brain tumors. [21] Immunohistochemical profile of PGNT complied from our case and reviewed cases shows a large array of positive expression of glial, neuronal, epithelial, and mesenchymal markers [Table - 1]. Differential diagnosis in our case included myxopapillary ependymoma, papillary tumor of the pineal region, and ganglioglioma. Myxopapillary ependymoma can be differentiated by the presence of PAS positive mucin cores around blood vessels, focal weak GFAP expression, and lack of immunostaining for neuronal markers. Papillary tumor of the pineal region is another newly described tumor entity that shows an epithelial-like growth pattern, in which the vessels are covered by a layer of large, columnar or cuboidal, tumor cells and nuclei are at the basal pole of tumor cells. Immunohistochemically, the tumor cells show strong staining for cytokeratin, S-100 protein, NSE, and vimentin but only weak or no staining for EMA and GFAP. [15] Ganglioglioma shows a lack of pseudopapillary pattern. Electron microscopic studies show the proliferating cells with a high nucleocytoplasmic ratio with scant cytoplasm. The cytoplasmic processes include both microfilaments (glial processes) and microtubules (primitive neurites) with membrane-bound granules of a neurosecretory type in the neuritic processes. [19] Genetic studies by Faria et al., in a single case of PGNT, revealed that the tumor was characterized by gains and structural alterations involving only chromosome 7 with breakpoints at 7p22 and a high-level amplification region at 7p14,q12. EGFR gene was not amplified. Recurrent rearrangements of chromosome 7 have been noted in other mixed glioneuronal tumors. The data in this case suggest that genes located at chromosome 7 can also be involved in the pathogenesis of PGNT. [22] The low proliferation index reported in PGNT is paralleled by a favorable clinical outcome. Most studies have reported a low MIB staining (< 3%). PCNA labeling index in our case was also negligible (< 1%). Mitotic activity was also not evident. A maximum uneventful follow up after surgery and radiotherapy of 17 years has been reported. [6] Complete/near complete resection is the treatment of choice. Radiotherapy and chemotherapy are withheld in lesions amenable to surgical resection. Tumor progression was minimal in our cases in a three month follow up. However, PGNTs do not always behave in a strictly benign fashion. Atri et al., have documented a high MIB labeling index of 11%. [1] Ishizawa et al. have also documented a case with oligodendroglial component with minigemistocytic change and high MIB labeling index. [9] Newton et al. [18] have described a case with high Ki67 index of 26 along with foci of necrosis and regions of mitoses. Two cases of aggressive PGNT with documented recurrence, local dissemination, and spread have been reported by Javahery et al. [19] Aggressive PGNT has also been treated with resection followed by radiation and temozolamide therapy. [18],[19] Postirradiation follow up MRI scans demonstrated a reduction of residual enhancement and FLAIR abnormality. [18] References
Copyright 2009 - Neurology India The following images related to this document are available:Photo images[ni09221t1.jpg] [ni09221f1.jpg] [ni09221f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}