 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Neurology India, Vol. 58, No. 1, January-February, 2010, pp. 99-102 Brief Report A rapid polymerase chain reaction-based test for screening Steinert's disease (DM1) Khalil Hamzi, Hanane Bellayou, Ilham Slassi, 1 Sellama Nadifi Medical Genetic Laboratory and Molecular Pathology, Medical School/Casablanca Morocco, 1 Neurology Department, CHU Ibn Rochd, Casablanca, Morocco Correspondence Address: Dr. Sellama Nadifi, Laboratory of Genetics and Molecular Pathology Faculty de Medicine and Pharmacy 19th , Tarik Ibn Ziad Casablanca, Morocco, labgenmed@yahoo.fr Date of Acceptance: 05-Jul-2009
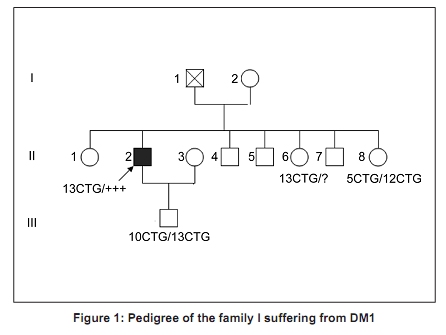
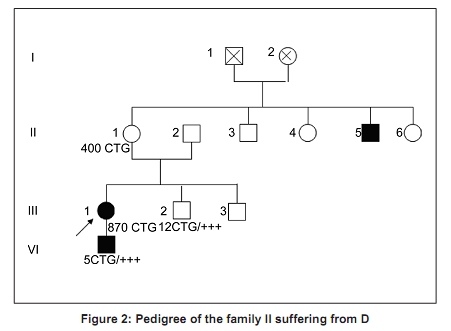
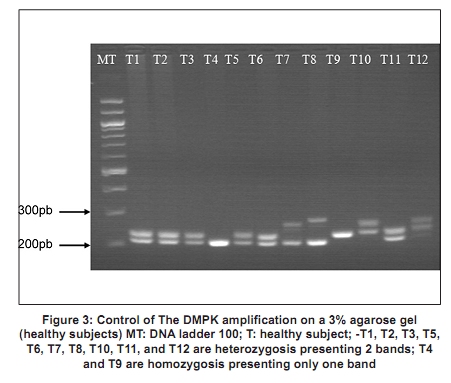
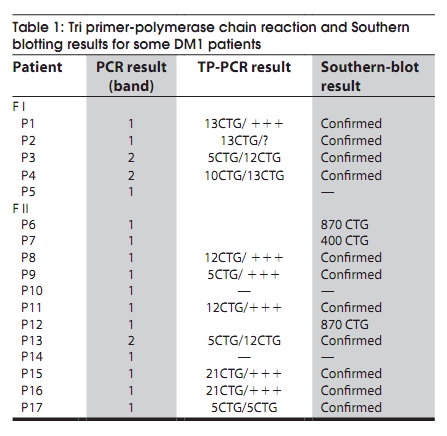
Code Number: ni10020 DOI: 10.4103/0028-3886.60411 Abstract Myotonic dystrophy (DM) is a multisystemic neuromuscular disorder caused by a dynamic mutation of (CTG) trinucleotide repeats in the 3' untranslated region of the myotonic dystrophy protein kinase gene (DMPK). The aim of the present study was to establish the use of polymerase chain reaction (PCR)-based simple and rapid method for initial sample screening. Only a minority of samples were tested positive with the above method and need to be detected by tri primer (TP)-PCR and Southern blotting which is more time consuming and involves use of radioactive material. This study concerned 24 patients from nine families with a clinical diagnosis of the DM1. DNA extracted from the blood was used for amplification of the triplet repeat sequences at the DMPK loci. We obtained two bands for the normal subjects and one band for patients corresponding to normal DMPK allele, confirmed by the TP-PCR and the Southern blot. This rapid test for initial screening of samples for the presence of DMPK mutations is economical and reliable method. This method reduces the number of samples needing TP-PCR and Southern blotting.Keywords: DM1, polymerase chain reaction, steinert′s disease, trinucleotide repeats CTG Introduction Myotonic dystrophy (DM) is a multisystemic neuromuscular disorder caused by a dynamic mutation of (CTG) trinucleotide repeats in the 3′ untranslated region of the myotonic dystrophy protein kinase gene (DMPK). [1] The number of CTG repeats observed in normal chromosomes varies from 5 to 37. The high variability in the size of expanded CTG alleles is associated with disparity in the ages of DM onset and severity of symptoms (anticipation phenomenon). [2],[3] Direct analysis of the CTG expansion mutation by Southern blotting is the laboratory method of confirmation of DM1 diagnosis. [4],[5] However, blotting is a relatively expensive and labor intensive procedure, particularly in the context of screening routine referrals for DM. So polymerase chain reaction (PCR) method has been suggested as an ideal alternative technique for screening the referrals then only the seemingly positive cases should be confirmed by Southern blotting method. The aim of this study was to establish the use of PCR-based method for initial screening of DM. [1] In this study we screened 24 DM patients using PCR method to detect CTG expansion and 30 healthy persons to evaluate our technique. Materials and Methods Twenty four patients from of nine families with DM referred for genetic consultation from the neurological department. For each patient a detailed family history was noted and a pedigree was drawn. All patients were examined in detail and a clinical scoring for DM1 was done. The family was counseled about the genetic testing and an informed consent was obtained. Blood sample of 5 ml was collected in EDTA and DNA was extracted using standard phenol-chloroform extraction protocol. [6] The following molecular genetic studies were done . We also studied 30 healthy subjects randomly selected from the general population to evaluate our method. Amplification for CTG repeats Amplification of DMPK triplet repeats was carried out using 100 ng of DNA in 25 µl reaction using 2 pairs of primers, 20 pmole of 5′-GAA GGG TCC TTC TAG CCG GGAA-3′ and 5′-CAG AGC AGG GCG TCA TGC ACA -3′ for amplication of DMPK CTG repeats as described previously. Taq DNA polymerase buffer (50.25 mmol/1 Tris-HCI, pH 8.8, 12, 45 mmol/L (NH 4 ) 2 SO 4 , 1 mmol/L of Mgcl 2 , and 127.5 ug/ml and 200 umol/L of dNTP. Hot start procedure was followed after denaturation at 95°C for 10 minutes, 2.5 U of Taq DNA polymerase was added to each tube white maintaining the temperature at 95°C for 1 minutes, 64°C for 1.5 minutes, 72°C for 2 minutes, with a final extension of 5 mintues at 72°C. The products were analyzed by electrophoresis through 3% agarose gel in an ethidium bromide stained. Results All the 24 patients were from Morocco. There were 14 males and 10 females and the age range of the subjects was 6 months-50 years. In [Figure - 1] and [Figure - 2] we present two examples of our nine pedigrees with a positive family history. Molecular Results Normal subjects In 30 subjects from the general population, we found a size of bands less than 310 bp (corresponding to a number of CTG less than 37), two bands for the heterozygous, and one intense band for the homozygous (T4, T9). [Figure - 3] shows amplification products from the DMPK loci analyzed on a 3% agarose gel for the control groups. The range of the PCR product of the normal DMPK allele varied from 205 to 310 bp for a number of repeats from 5 to 37. The subjects T1, T2, T3, T5, T6, T7, T8, T10, T11, and T12 are heterozygous presenting two bands corresponding to two numbers of triplets, T4 and T9 are homozygous presenting only one band. This number is heterogenic between subjects and always less than 37. Patients All the 24 patients were screened for DMPK mutations by PCR approach. [Figure - 4] shows result for patients. TP-PCR and Southern blotting were done for confirmation and the results were similar as presented in [Table - 1]. Discussion and Conclusion DM is the most common inherited neuromuscular disease with a worldwide prevalence of 2.1 to 14.3 per 100,000 inhabitants and incidence of 1 in 8,000 births. [7] It is a progressive multisystemic autosomal dominant disorder with phenomena of anticipation, it denotes progressively earlier onset of the disease with in successive generations. [8] The disabilities are substantial and therefore early detection is mandatory for reproductive counseling of families in which the DM1 has been observed. In molecular methods, Southern blotting, although used widely has a long laboratory turn around time, is relatively expensive and has got hazards associated with the use of radioisotopes. [9] So PCR method is a rapid and cheap method for initial screening. The best protocol would be to perform PCR for initial screening followed by TP-PCR and Southern blots for confirmation. We sought the number of repetitions in 24 patients referred from the department of neurology, first by PCR standard then by TP-PCR and Southern blot to be able to compare the results of different techniques. The standard PCR allowed us to amplify the normal allele (smaller than 310 pb) among witnesses to the general population, we were able to obtain either two bands of distinct size < 310pb or an intense band which corresponds to two overlapping bands of the same size. In patients we have obtained only one band corresponding to the normal allele, PCR could not amplify the pathological fragment causes its large size (high number of repetitions CTG). The TP-PCR can detect the presence of long allele size without determining the total size of the expansion or the exact number of expansions CTG. [10] Our results are consistent with those of Warner et al. on a comparative study of TP-PCR and the Southern-blot in detecting mutation unstable; PCR fluorescent reduces the use of Southern-blot allowing the identification of the expansion (presence of an abnormal number of repetitions), but the Southern-blot is still required if an accurate estimate of the size and number of triplets is sought. [10],[11] The results of TP-PCR and Southern-blot confirmed the results of our method which could amplify the normal allele which is logical, since the Taq polymerase can not exceed 1000 bp and that the mutated alleles identified by Southern blot-CTG are 400 and 870 CTG which corresponds to sizes of 1,399 bp and 2,809 bp, respectively. Regarding patients who have found no mutation in the PCR, confirmed by the absence of mutation in TP-PCR and Southern-blot, it may be another muscular dystrophy mainly DM 2 or proximal myotonic myopathy (PROMM) disease caused by an expansion of tetranucléotides (CCTG) at the ZNF9 gene (zinc finger protein). [12],[13],[14] We conclude that PCR based screening for DM1 is reliable and should be used as an initial screening test for all patients with DM and only the seemingly positive cases should be confirmed by Southern blotting method. Acknowledgments We are deeply grateful to the nine families for their invaluable collaboration to this study. We are also grateful Mr. Pierre Ray of genetic molecular laboratory, Grenoble; Mr. Pierre Boisseau of genetic molecular laboratory, Nantes, for accepting the confirmation of our results by screening with twice TP-PCR and southern-blot methods. References
Copyright 2010 - Neurology India The following images related to this document are available:Photo images[ni10020f4.jpg] [ni10020f2.jpg] [ni10020f1.jpg] [ni10020t1.jpg] [ni10020f3.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}