 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
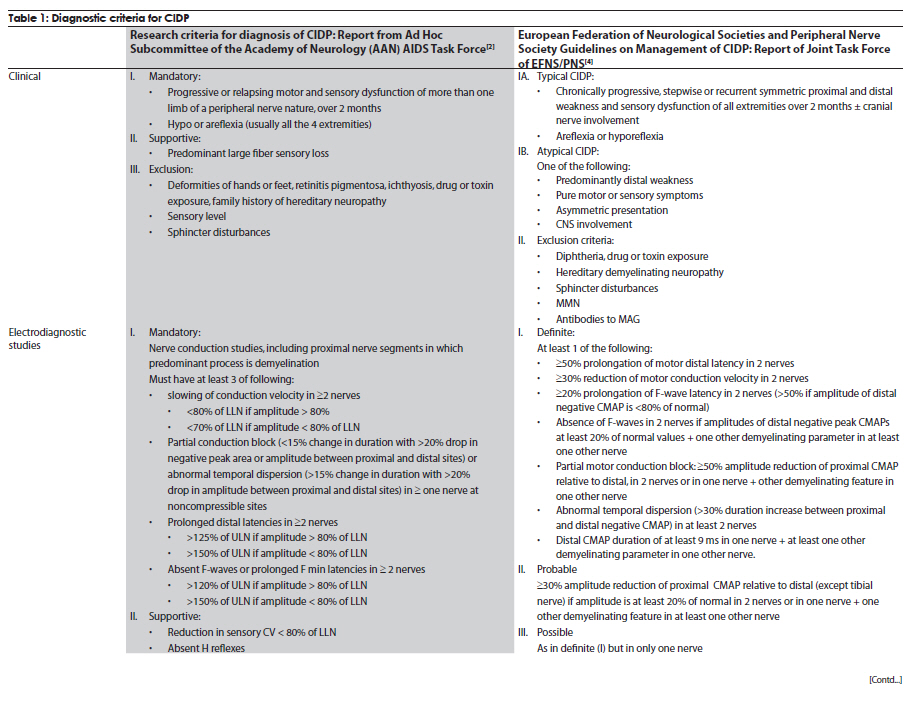
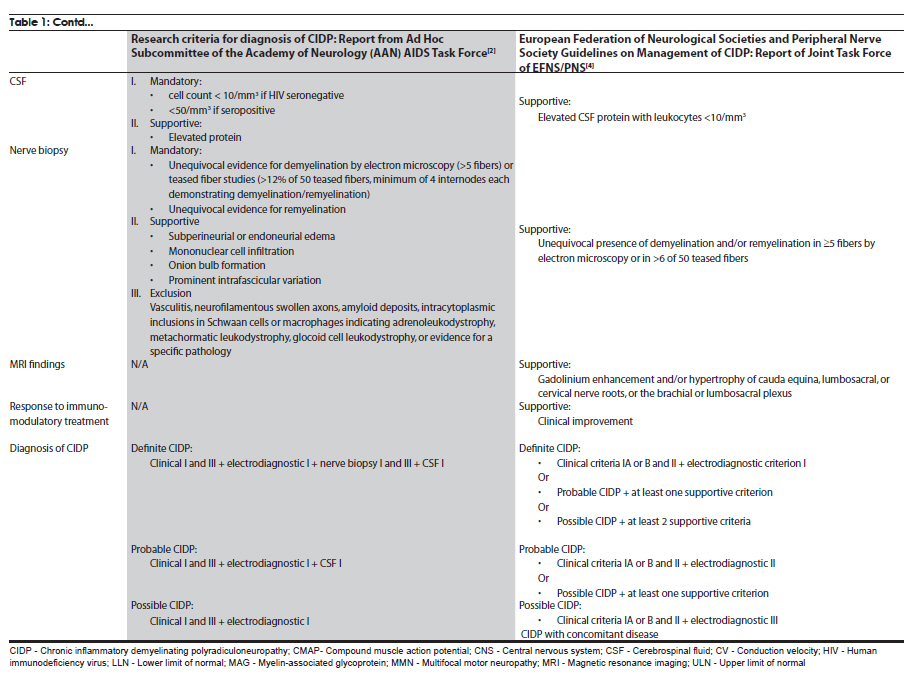
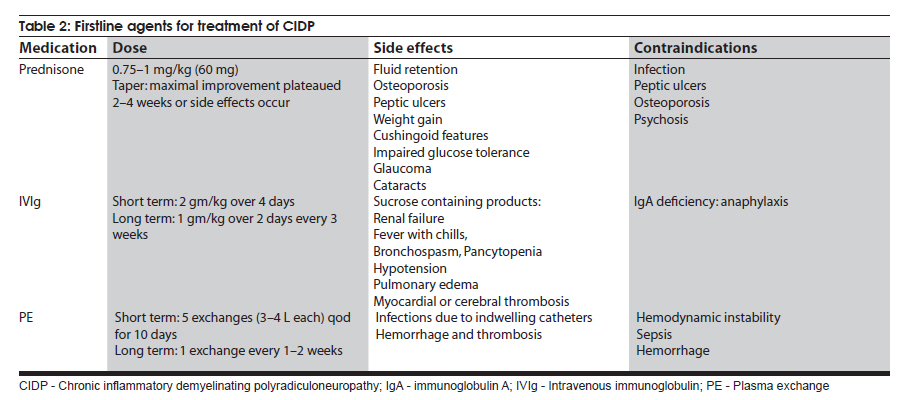
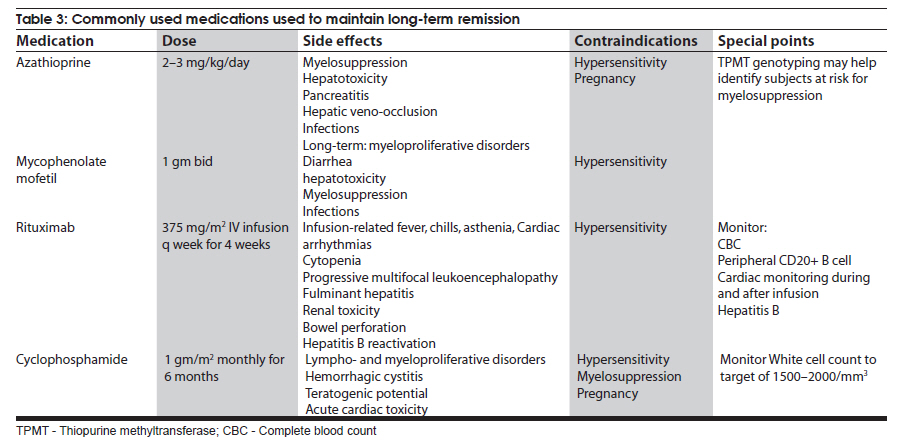
Neurology India, Vol. 58, No. 3, May-June, 2010, pp. 351-360 Review Article Management strategies in chronic inflammatory demyelinating polyradiculoneuropathy Kamakshi Patel1, Minal Bhanushali2, Suraj Ashok Muley3 1 Department of Neurology and Ophthalmology, Michigan State University, East Lansing, Michigan, USA Date of Acceptance: 17-Jun-2010 Code Number: ni10097 PMID: 20644261 Abstract Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) is a chronic, proximal and distal, asymmetrical or symmetrical, motor and sensory demyelinating polyneuropathy with a progressive course for at least 2 months. The accurate diagnosis is crucial as CIDP is amenable to treatment. Recent advances have provided new strategies and options for management of this syndrome. In this article, we review the clinical and diagnostic features as well as discuss recent insights and treatment strategies along with our experience in the management of patients with CIDP. Keywords: Chronic demyelinating neuropathy, chronic inflammatory demyelinating neuropathy, variants of chronic demyelinating neuropathy Introduction Chronic inflammatory demyelinating polyradiculo-neuropathy (CIDP) is a sensorimotor neuropathy that evolves over at least 2 months with either a progressive or a relapsing remitting course. [1],[2] Variants of CIDP with distinct clinical presentations have been described and their recognition is important because of varied treatment responses. In general, patients with CIDP present with acute to subacute onset of proximal and distal weakness, and sensory symptoms that have a progressive or stepwise course for over 2 months. [1],[2] About 15% of patients have an acute onset that can be confused with Guillain-Barrι syndrome (GBS) and the diagnosis of CIDP is made only in retrospect. [3] Physical examination shows hyporeflexia or areflexia that is out of proportion to the degree of weakness, suggesting that the underlying pathology is that of segmental demyelination. [2],[4] Various diagnostic criteria have been proposed for CIDP. These criteria vary in their specificity and sensitivity. The American Academy of Neurology (AAN) criteria were initially proposed in 1991 mainly for research purposes and had a specificity of 100% [2],[5] but lacked sensitivity (45.7%) and were found to be inadequate for routine clinical practice. The recently proposed European Federation of Neurological Societies and Peripheral Nerve Society (EFNS/PNS) guidelines on management of CIDP [4] provide diagnostic criteria that may have greater clinical applicability and improved sensitivity of 81.3% and specificity of 96.2% compared with the AAN criteria. The EFNS/PNS guidelines have taken into consideration the cerebrospinal fluid (CSF) changes and response to immunomodulatory treatment. Diagnostic role of enhancing or enlarged nerve roots on neuroimaging were also incorporated, but the role of nerve biopsy remained limited [4] [Table - 1a and Table - 1b]. There are numerous other criteria proposed, however, [6],[7],[8] the discussion of those is beyond the scope of this article. Nevertheless, clinical experience of neurologists world over suggests that there are a significant number of patients who do not satisfy these diagnostic criteria but still respond to treatments that are effective for CIDP. [9] Thus it is prudent to consider the diagnostic possibility of CIDP when the history, physical examination, and electrodiagnostic studies suggest an acquired demyelinating neuropathy. Chronic inflammatory demyelinating polyradiculoneuropathy-variants Over the last few decades, variations in the clinical and electrodiagnostic features of CIDP have been described. These diseases have a common denominator in that all are related to focal segmental demyelination but have distinctive treatment responsiveness, suggesting varied underlying pathophysiologic mechanisms. Distal acquired demyelinating symmetric neuropathy Patients with distal acquired demyelinating symmetric neuropathy have a slowly evolving, sensory predominant larger fiber neuropathy with relative preservation of motor function that typically presents with sensory ataxia. A significant proportion (66%) of these patients have IgM paraproteinemia and/or antibodies to myelin-associated glycoprotein (MAG). [10] Distally accentuated conduction slowing is the electrophysiologic hallmark of these neuropathies. [11] It is important to rule out myeloproliferative disorders in these patients. Recognition of this disorder is important since response to treatment is different from that in CIDP. Multifocal motor neuropathy Patients with multifocal motor neuropathy (MMN) present with focal weakness in the distribution of individual peripheral nerves without sensory symptoms. Electrodiagnostic testing reveals partial conduction block related to segmental demyelination with sparing of the sensory responses even through the site of conduction block. [12] In some patients, who are otherwise indistinguishable from the prototypic patients, conduction block may not be demonstrable with routine electrodiagnostic testing. [13],[14] Anti-GM1 antibodies are seen in a proportion of these patients but are not required for the diagnosis. [15] This disorder can be easily confused with motor neuron disease because of the lack of sensory findings, but careful clinical examination that reveals weakness in a peripheral nerve rather than segmental distribution and a high index of suspicion can aid in the diagnosis. Recognition is important since these patients may respond to immunomodulatory treatments. Multifocal acquired demyelinating sensory and motor neuropathy: Lewis-Sumner syndrome These patients present with weakness and numbness in the distribution of individual peripheral nerves related to focal demyelinative conduction block. [16] Some of these patients can evolve to develop diffuse confluent involvement that can be indistinguishable from CIDP. [17] Given that response to treatment may be similar to that seen with CIDP, there is some controversy as to whether this is a distinct pathophysiologic entity or whether it belongs to the same spectrum as CIDP. Diagnosis Diagnosis of CIDP is primarily made on the basis of clinical presentation and electrodiagnostic findings. A clinical picture of progressive weakness and numbness over a duration of 8 weeks with hyporeflexia that is out of proportion to the degree of weakness is characteristic. [2],[4] The presence of proximal weakness in a neuropathy suggests involvement of nerve roots and is common in patients with CIDP. [1] Electrodiagnostic findings are also important diagnostic tools and show incongruous slowing of conduction velocities related to patchy segmental demyelination and remyelination. [2] Typical findings on nerve conduction studies include at least 20-30% slowing of conduction velocities (compared with lower limit of normal [LLN]) at sites that are not prone to entrapment, 125%-150% (compared with LLN) prolongation of distal latencies and F-wave latencies. [2] Segmental amplitude change of 20-50% in the compound muscle action potential related to partial conduction block and/ or abnormal temporal dispersion (>15% change in duration between proximal and distal sites can also be seen. [2],[4] Conduction block is more common in patients with MMN and Lewis-Sumner syndrome (LSS). [14],[16] Reduction in the motor and sensory amplitudes are also seen and are commonly related to secondary axonal degeneration but can also be related to very distal conduction block. [18] Electromyography though not included in the diagnostic criteria, if performed shows findings that are consistent with acute and/or chronic partial denervation but reduced recruitment that cannot be explained on the basis of axon loss suggests the possibility of conduction block that may be proximal and not easily evident on nerve conduction studies. [19] CSF analysis can show cytoalbuminogenic dissociation with elevated protein levels without elevation in the white cell count. When elevation of the white cell count is seen human immunodeficiency virus (HIV) infection should be considered. [2] When CSF analysis is done early on in the disease process, elevation of protein level may not be seen. [20] Rarely, patients with idiopathic CIDP can have CSF pleocytosis. [20] The role of nerve biopsy in the diagnosis of CIDP has been diminishing. Several studies have shown that the nerve biopsy histopathology is not significantly different in patients with axonal neuropathy compared with CIDP. [5],[21] We reserve nerve biopsy only for patients with atypical clinical presentation where the diagnosis is not obvious with routine testing. Other diagnostic workup to exclude concomitant diseases include complete blood counts and erythrocyte sedimentation rate, hepatic and renal function tests, antinuclear antibody, extractable nuclear antigen autoantibodies, thyroid function tests, glucose tolerance test, serum and urine immunofixation or immunoelectrophoresis, HIV and hepatitis serology, antibodies to Borrelia burgdorferi or Lyme disease, and angiotensin I-convering enzyme levels. [2] Chest radiograph and in patients unresponsive to treatment, a skeletal survey may be repeated. [4] Hereditary neuropathy may be excluded with examination of the family members or performing analysis for PMP22 deletion or duplication. Management Goals of treatment Effective treatment of CIDP is predicated on early diagnosis when the degree of axon loss is relatively mild. Once there is significant secondary axon loss, response to treatment can be incomplete. The goals of treatment are to improve weakness, prevent disability, and induce and sustain a remission. It is important to achieve these goals in a cost-effective manner with minimal treatment-related adverse effects. General approach The firstline treatments for CIDP include prednisone, intravenous immunoglobulin (IVIg), and plasma exchange (PE) [Table - 2], [Figure - 1]. These treatments have been shown to be effective, but whether one treatment is better than the others (in terms of improving weakness and inducing a remission) remains unclear. Although IVIg and PE have been shown to be effective in placebo-controlled trials, it remains unclear whether these treatments are effective in inducing a long-term remission. [22] Prednisone has also been shown to be effective in improving weakness but long-term treatment that is often necessary in CIDP results in severe steroids-related adverse effects. Newer treatment regimens that use high-dose pulsed steroids may be better tolerated and perhaps more effective in improving weakness and inducing remission but need further study. [23],[24] Long-term immunosuppressants, such as azathioprine and mycophenolate mofetil, can be used, when the primary treatment is not sufficient and as steroid or IVIg-sparing agents. In intractable cases, cyclophosphamide with or without stem cell rescue has been shown to be effective. [25],[26],[27] These treatment options have been outlined in [Table - 2] and [Table - 3]. We typically start treatment with corticosteroids as they are proven to be cost effective and clinically efficacious in CIDP treatment, through long-term clinical experience with its use. [28] The authors typically start with pulsed steroids administered in a once-a-week regimen. [24] In patients who have no response to treatment within 1-3 months [28] or unable to tolerate steroid-related adverse effects, we would switch to IVIg. [4] Daily prednisone, IVIg, or PE can also be used as firstline treatments but there is a subset of patients who may not respond to IVIg because of genetic predisposition. [29] In general, about 66% of patients respond to one of the firstline treatments (corticosteroids, IVIg, or PE). [30] Once adequate improvement is accomplished, a slow taper of the primary therapy is instituted with close surveillance to monitor for relapse. We use a 120-point neuromuscular scale to monitor strength and Inflammatory Neuropathy Cause and Treatment (INCAT) disability scale to monitor function. [24] Long-term immunosuppressants can be used if there is difficulty with the taper or if response to treatment is suboptimal. It is important to remember that a lack of complete resolution of weakness may be related to axonal degeneration, which may not respond to more aggressive immunotherapy and may improve over time with neural repair mechanisms. Individual therapies Glucocorticoids The efficacy of corticosteroids for treatment of CIDP has been described in only one randomized study. [31] In this study, some improvement in strength and other objective measures was observed compared with patients who received no treatment. Subsequently, other studies have compared corticosteroids with other treatment modalities or have used steroids in conjunction with other agents. [7] Forms and dosing: The randomized study [32] used prednisone in the dose of 120 mg every other day, with subsequent tapering of the dose to 0 mg over 12 weeks, and there was improvement in 86% of treated patients. The study by Hughes et al., [7] used prednisone 60 mg/day with tapering of the dose to 10 mg over six weeks. [7] Other studies have typically used prednisone in doses of 40-100 mg for 2-4 weeks, followed by a gradual or alternate day taper. [32],[33] Molenaar and colleagues [34] used pulsed high-dose dexamethasone, 40 mg daily for 4 days every 28 days for 6 months, in CIDP and had shown a 70% response rate with fewer adverse effects. However, pulsed high-dose dexamethasone treatment did not induce remission more often than prednisone treatment in pulsed high-dose dexamethasone vs standard prednisone treatment for CIDP (PREDICT) study. High-dose dexamethasone could be considered as induction therapy in CIDP, but comparison with IVIg treatment is needed. [23] A retrospective study showed that intravenous methylprednisone in doses of 1000 mg IV daily for 3-5 days, followed by once a week for 1 month, and later once every 2-12 weeks was as effective as IVIg and oral prednisone, but with fewer side effects from corticosteroids compared with oral prednisone. [35] Oral methylprednisone at a dose of 500 mg once a week had shown improvement in the majority of patients with a remission rate of 60% that was sustained over several years; the treatment was well tolerated in most patients with a syndrome of unpleasant taste and insomnia lasting for up to 2 days after each dose. [24] Corticosteroids are not useful in certain forms of CIDP, such as purely motor CIDP and MMN, [36],[37] and may actually worsen the weakness. Intravenous immunoglobulin IVIg was approved for treatment of CIDP in the United States in 2008. There have been 7 randomized controlled trials, involving 287 patients, that have proven the efficacy of IVIg. [22],[38],[39],[40],[41],[42] The IVIg for CIDP (ICE) trial [39] was a double-blinded randomized, placebo-controlled trial involving 117 patients with a crossover design. The study showed that 54% of patients treated with IVIg as opposed to 21% of patients receiving placebo showed improvement in adjusted INCAT disability score that was maintained during the crossover time. There is a 0-3% risk of thromboembolic events, such as stroke, myocardial infarction, pulmonary embolism, in patients receiving IVIg. [43] Renal failure may occur as a complication of older preparations containing high osmolarity sucrose solvent. [44] Patients with preexisting renal disease, diabetes mellitus, hypovolumia, sepsis, concomitant use of nephrotoxic drugs, and age more than 65 years have been identified as risk factors for developing nephrotoxicity. [45] Checking renal function prior to infusion and thereafter has been recommended by the Center for Disease Control and Prevention (CDC). [45] However, this adverse effect has been minimized with use of newer sucrose-free preparations. Reversible vasospasm, headaches, rash, congestive heart failure, transient drop in blood cell counts, [46] and the theoretic risk of transmission of infectious diseases are some of the other rare adverse events reported with the use of IVIg. Plasma exchange PE is effective in CIDP and this has been demonstrated in 2 randomized controlled trials involving 47 patients. [47],[48] Adverse effects, such as infection from indwelling catheter, pneumothorax, vessel perforation, hypotension, electrolyte imbalance, citrate-induced hypocalcemia, thrombosis, and bleeding are seen in 3-17% of patients. [49] Patients with MMN may show deterioration with PE. [50] Long-term immunosuppressants Steroid sparing agents commonly used to maintain remission include azathioprine and mycophenolate mofetil. Mycophenolate mofetil was shown to be effective in reducing pain or showing minimal improvement in disability scales in an open-label study. [51] Another retrospective study found modest benefit in about 20% of patients and stabilized patient condition, allowing reduction of steroid or IVIg therapy. [52] There have been small numbers of nonrandomized, open-label studies showing efficacy of other agents, such as cyclosporine, fludarabine, interferons, and etanercept. Intractable and refractory cases may respond to cyclophosphamide and rituximab. However, there is only one double-blinded placebo-controlled study assessing the efficacy of rituximab in patients with anti-MAG demyelinating neuropathy. There was 34% reduction in IgM, 50% reduction in anti-MAG titers, and improvement in INCAT scores in 4 of the 13 patients receiving rituximab. [53] Rituximab may also be effective in CIDP patients associated with thrombocytopenia, [54] diabetes, [55] anti-SGPG IgM antibody, [56] and Evans syndrome. [57] Similarly, there has been a small study reporting the use of cyclophosphamide in conjunction with PE, to be effective in patients with MMN [58] and anti-MAG [59] neuropathy. In a single patient with IVIg-dependent relapsing CIDP, unresponsive to steroids or conventional immunosuppressive agents, remission was achieved following treatment with alemtuzumab. [60] Treatment of specific forms of CIDP MMN: IVIg is the firstline treatment for patients with MMN. [61],[62] Plasmapheresis and corticosteroids have been associated with worsening in these patients, and should be avoided. [35],[36] For patients resistant to IVIg, secondline treatments, such as chemotherapeutic agents are used. Anti-MAG neuropathy: Anti-MAG neuropathy has a relatively benign course, and treatment is not warranted for most of the patients. However, when treatment is warranted, rituximab is the treatment of choice as it reduces the levels of IgM. [53] LSS: Patients with LSS may respond to both steroids and IVIg. We typically start treatment with steroids and reserve IVIg for patients who are either unresponsive to treatment or develop intolerable adverse effects. Sensory variant of CIDP: The treatment for CIDP with sensory predominance is same as in CIDP. However, considering the risks involved in CIDP treatment, we defer treatment only if there is sensory ataxia, or the weakness is severe enough to impair activities of daily living. Acute presentation of CIDP: About 15% of CIDP patients present with rapidly progressive symptoms. [3] During this time, the clinical picture is indistinguishable from GBS, and the diagnosis is often made only in retrospect. The symptoms continue to progress over 8 weeks in CIDP, whereas they tend to plateau by 4 weeks in GBS. During the acute phase, management consists IVIg and PE. CIDP with central nervous system (CNS) involvement: There have been several reports suggesting subclinical involvement of the CNS in patients with CIDP. [63],[64],[65],[66],[67] CNS involvement is evidenced by MRI findings and evoked potential testing. Typical lesions of multiple sclerosis (MS) are infrequent in patients with CIDP. [67] The treatment is no different with or without CNS involvement. There is a suggestion that CIDP in patients with CNS involvement is more demyelinating, and hence more responsive to immunotherapy compared with CIDP without CNS involvement that can have axonal damage as well. [64] If clinical presentation warrants, MS or other inflammatory causes of white matter lesions need to be ruled out. Diabetes and CIDP: There has been suggestion that CIDP is more prevalent in patients with diabetes, [68] although a recent study suggested that the incidence of diabetes was lower in patients with CIDP than in healthy controls. [69] The reported higher incidence of CIDP in diabetes has been attributed to misdiagnosis since conduction slowing is commonly seen in diabetes and overreliance on electrodiagnostic features can be misleading. In our experience, there are rare instances of the coexistence of diabetes and CIDP and in these cases the pathogenic relation between CIDP and diabetes remains unclear. We would consider a diagnosis of CIDP in patients with diabetes if there is progressive proximal and distal weakness, abnormal temporal dispersion, or conduction block at nonentrapment sites on electrophysiologic studies and markedly elevated CSF protein levels. It has been shown that CIDP patients with diabetes have a higher frequency of autonomic dysfunction, unrecordable motor, sensory nerve conductions and F-waves, and reduced compound muscle action potential. Also, the degree of conduction block is higher in CIDP patients without diabetes, than those with CIDP and diabetes. [70] In most instances, definitive and objective response to immunotherapy is used to solidify the diagnosis. We typically start treatment with pulsed oral prednisone. In our experience, despite elevation of blood glucose levels for 1-2 days after each dose, the treatment is well tolerated. In patients who develop intolerable adverse effects with this regimen or in patients who do not respond to treatment, we use IVIg. Close monitoring for renal failure is necessary since diabetics commonly have subclinical nephropathy and IVIg can precipitate renal failure in these patients. Long-term treatment strategies are similar to those for idiopathic CIDP. There has been one study that used SQIg in 2 patients with CIDP, and showed benefit with IVIg. The use of SCIg was well tolerated, and led to stabilization of their disease course. [71] Further controlled studies are required to prove the efficacy of this form of treatment, as it would be ideal for diabetic patients who develop CIDP. References
Copyright 2010 - Neurology India The following images related to this document are available:Photo images[ni10097t2.jpg] [ni10097t3.jpg] [ni10097t1b.jpg] [ni10097f1.jpg] [ni10097t1a.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}