 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
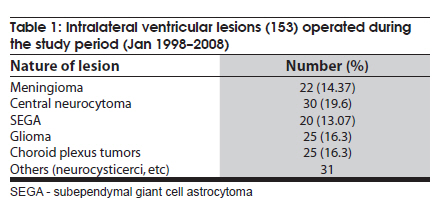
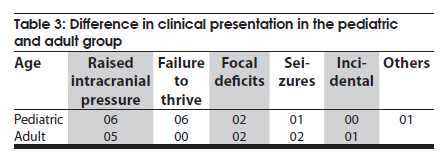
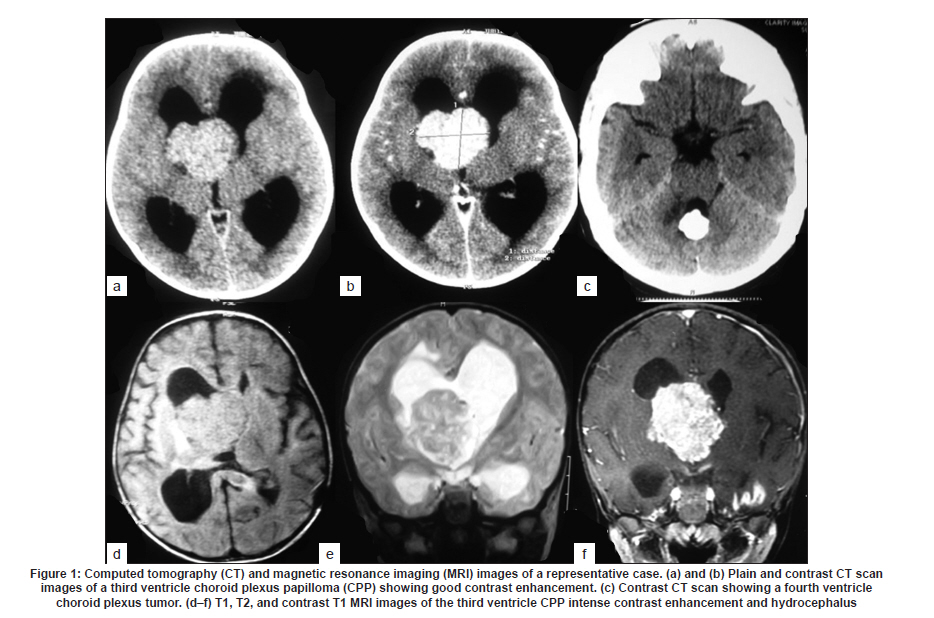
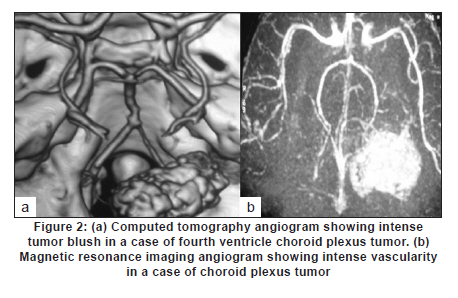
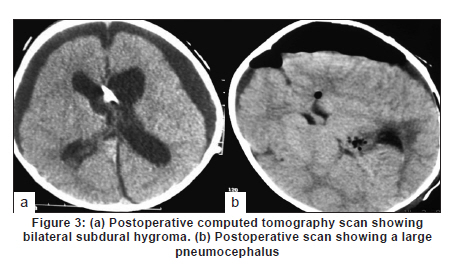
Neurology India, Vol. 58, No. 3, May-June, 2010, pp. 429-435 Original Article Choroid plexus tumors: An institutional series of 25 patients Girish Menon, Suresh N Nair, Sachin S Baldawa, Ravimohan B Rao, KP Krishnakumar, CV Gopalakrishnan Department of Neurosurgery, Sree Chitra Tirunal Institute for Medical Sciences and Technology, Trivandrum, India Date of Acceptance: 11-Jun-2010 Code Number: ni10109 PMID: 20644273 Abstract Purpose : Choroid plexus tumors (CPT) are rare neoplasms that pose considerable treatment challenges. This study reviews a single institute's experience with 25 patients of CPT and attempts to contribute to the general body of knowledge on CPT. Keywords: Carcinoma, choroid plexus, papilloma, ventricle Introduction Choroid plexus tumors (CPTs) are uncommon neoplasms derived from the choroid plexus epithelium and characterized by papillary and intraventricular growth. CPTs pose formidable treatment challenges. The young age at presentation, controversies surrounding the pathologic classification of these tumors, high risk of surgical morbidity, and limited data concerning the effectiveness of adjuvant therapy contribute to the management dilemmas. We reviewed our experience of having treated 25 patients with CPT with an intention to contribute to the general body of knowledge of these tumors. Materials and Methods We retrospectively analyzed the case records of all the patients operated for CPTs since January 1998 and having a minimum of one year follow up. The variables analyzed include age, sex, presentation, imaging characteristics, extent of resection, histopathology, recurrence and role of adjuvant therapy. Glasgow outcome score was used to analyse outcome. Results CPTs constituted 0.52% (25/4849) of the total number of brain tumors and 16.3% (25/153) of the lateral intraventricular tumors operated in our institute during the study period [Table - 1]. A definite male preponderance was observed in our series with a male to female ratio of nearly 2:1. The mean age of patients with choroid plexus papilloma (CPP) was 18.25 years (range, 6 months to 50 years; SD, 21.8), of patients with choroid plexus carcinoma (CPC) was 17.5 years (range, 10 months to 37 years; SD, 17.46), and that of patients with atypical CPP was 22.1 years (range, 9 months to 43 years; SD, 24.1) [Table - 2]. The tumors were distributed as follows: lateral ventricle (16; 64%), fourth ventricle (5; 20%), fourth ventricle with cerebellopontine angle extension (3; 12%), and third ventricle (1; 4%). All the 13 patients younger than 14 years had supratentorial CPTs, whereas 8 of 12 patients above 14 years had infratentorial CPTs. For the 16 lateral ventricle CPTs, the mean age was 7.8 years, whereas for the 8 infratentorial CPPs the mean age was 41.6 years. The single patient with CPP in the third ventricle was aged 2 years. In the pediatric group (age < 14 years, n = 13), the chief presenting symptoms were raised intracranial pressure headache and delayed milestones [Table - 3]. In one child the lesion was detected incidentally and another child presented with polyphagia and obesity. In the adult group (age > 14 years, n = 12), the chief presenting symptoms included raised intracranial pressure headache, seizures, and focal neurologic deficits [Table - 3]. Patients with CPT of the fourth ventricle had, in addition to features of raised pressure, cerebellar signs (11 cases), and patients with CPT of the cerebellopontine angle presented with cranial nerve palsies. Plain computed tomography (CT) scan findings are variable and the lesion appeared hyperdense in 12, isodense in 9, and hypodense in 4 [Figure - 1]a-c. Calcification was common and was seen in 12 (48%) of our patients. Magnetic resonance imaging (MRI) was performed in 18 patients. All the tumors were of intermediate signal intensity on T1-weighted images and of increased signal intensity on T2-weighted images. On contrast administration, all the lesions enhanced uniformly both on CT scan and MRI [Figure - 1]d-f. CT angiogram and MR angiogram revealed intense tumor blush distinguishing them from other intraventricular tumors [Figure - 2]a and b. The patients were operated through either parietal, suboccipital, or retromastoid craniotomy route depending on the location of the tumor. Based on the intraoperative impression and the postoperative CT scan, a total resection was achieved in 19 patients, near total excision in 3, and subtotal decompression in 3. The main reasons for the incomplete excision include tumor infiltration into the ependyma, poor plane of cleavage, and tumor vascularity. Histopathologic examination was suggestive of CPP in 12, CPC in 9, and atypical CPP in 4 [Table - 4]. Recurrence occurred in 4 patients, namely, 2 patients with atypical CPP and 1 patient each with CPP and CPC. The first patient was a 50-year-old woman with a CPP located in the fourth ventricle, who presented with a cystic recurrence 9 years after total excision. She did well after reoperation for the recurrent lesion. The second patient was a one-and-a-half-year-old child who underwent total resection of a histopathologically proven CPC in the right lateral ventricle followed by adjuvant chemotherapy, but developed recurrence 6 months later and underwent decompression followed by a repeat course of chemotherapy. The third patient, a 43-year-old woman who was initially diagnosed to have CPP of the fourth ventricle and had undergone near total excision, presented with recurrence in the third ventricle and suprasellar region 6 years after initial surgery, followed by drop metastasis in the lumbar subarachnoid space 2 years later. A review of earlier slides and histopathology at second surgery was suggestive of atypical CPP, and she underwent craniospinal irradiation. The fourth patient, a 43-year-old male underwent subtotal decompression of a cerebellopontine angle CPP initially. He developed recurrence in the middle cranial fossa 4 years later for which he underwent radical excision of the recurrence followed by 28 cycles of cranial radiotherapy. He developed drop metastasis in the lumbar space 2 years later and underwent lumbar decompression. A repeat histopathology was suggestive of atypical CPP, and he underwent craniospinal irradiation. Major postoperative complications included pneumocephalus (10/25; 40%) and subdural effusions (8/25; 32%) [Figure - 3]. Subdural effusions can be quite refractory to treatment and required a placement of a subduroperitoneal shunt in 2 patients). Four of our patients developed postoperative motor deficits and 5 developed transient dysphasia following surgery. One patient developed extradural hematoma requiring emergency evacuation. Twenty patients in our series had evidence of preoperative hydrocephalus of which 2 required ventriculoperitoneal (VP) shunts prior to definitive surgery and 4 required VP shunts after definitive surgery. The mean follow-up of patients with CPP was 5.4 years (range, 1-10 years). None of the patients with CPP received any form of adjuvant therapy even after incomplete excision. One patient had a recurrence, which was reoperated. All the patients were doing well at last follow-up and had a Glasgow Outcome Score (GOS) of 5. The median survival for patients with CPC who underwent gross total excision with adjuvant therapy was 58 months and those who had a subtotal resection with adjuvant therapy was 36 months. Of the 9 patients with CPC, 3 received both chemotherapy and radiotherapy, 2 received only radiotherapy (craniospinal irradiation), 1 received only chemotherapy, and 2 did not receive any adjuvant therapy. One patient died within a week after operation due to surgical complications, 1 died 2 years later due to widespread dissemination, and 2 other patients died on follow-up due to nonsurgical causes. One patient who was reoperated for a recurrence is moderately disabled but independent for activities of daily life. Two patients are doing well, but 2 patients with focal deficits were lost to follow-up after 1 year. The mean follow-up of patients with atypical CPP was 26 months (range: 18-44 months). Of the 4 patients with atypical CPP, 2 underwent repeat surgeries for recurrence followed by adjuvant chemotherapy and are in GOS 3 at last follow-up. The third patient was a 20-month-old boy who was too young for radiotherapy and his relatives were not willing for chemotherapy. He, however, is doing well without any deficits 2 years after surgery. The fourth patient was a 9-month-old girl who underwent chemotherapy for 6 months after surgery. She has also completed 2 years after surgery but still has a large head size. Repeat imaging shows bilateral subdural hygroma with no tumor recurrence or hydrocephalus. Discussion CPTs are rare childhood neoplasms that account for 0.3-0.7% of all intracranial tumors, 2-6% of pediatric brain tumors, and 10-20% of brain tumors in children younger than 1 year. [1],[2] Choroid plexus neoplasms arise wherever choroid plexus tissue exists; lateral ventricle is thus the most common site (50% of cases) for these tumors, followed by the fourth ventricle (40%), and the third ventricle (5-9%). [1],[3] Our findings are similar, with a majority (16; 64%) of the tumors being in the lateral ventricle, 5 (20%) in the fourth ventricle, 3 (12%) in the fourth ventricle with cerebellopontine angle extension, and 1 (4%) in the third ventricle. CPTs arising in the lateral ventricle are usually attached to the choroid plexus in the trigone region, whereas those located in the third ventricle have their attachment to its roof and fourth ventricular tumors are attached to the posterior medullary velum. [4] About 5% of CPTs are seen in more than one location and rarely CPTs are reported to arise from embryonic cell rests in extraventricular locations, including the cerebellopontine angle, the suprasellar region, the frontal lobe, the posterior commissure, the pineal gland, and the cerebellum. [5],[6],[7],[8],[9],[10],[11],[12],[13],[14],[15],[16] CPTs are known to manifest in the first decade of life, especially in the first 2 years. [7],[13] The typical age at presentation varies with the location of the tumor. Lateral ventricular tumors are much more common in patients aged 10 years or younger, whereas those that arise in the fourth ventricle are fairly evenly distributed among patients 0-50 years of age. The mean age at presentation in our series was 18.6 years (range, 6 months to 54 years). All patients younger than 14 years had supratentorial CPTs, whereas a majority (8/12) of patients older than 14 years had infratentorial CPTs. No gender predilection has been reported for lateral ventricular tumors, whereas those that occur in the fourth ventricle are more commonly found in males. [1] In our series, as in some other series, [11] however, a definite male preponderance was observed with a male to female ratio of 2:1. A variety of genetic loci are implicated in the development of CPTs and the loci associated with CPC generally differ from those associated with CPP, suggesting a separate genetic basis for these 2 lesions. Patients with CPPs have frequently displayed the following chromosomal additions and deletions: 17q; 15q; 17p; 15p; 19p; 19q; and 112p and 112q; 18q; 210q; 210p; and 222q. Patients with CPCs have primarily displayed the following additions and deletions: 112p; 112q, and 120p; 11, 14q, and 120q; 14p; 18q and 114q; 17q, 19p, and 121; 222q; 25q; 25p and 218q. Aicardi syndrome, Li-Fraumeni syndrome, constitutional 9p duplication, and in the setting of an X; 17(q12;p13) translocation, hypomelanosis of Ito are a few genetic abnormalities associated with the development of CPPs. Similarly, polyomaviruses SV40, JC, and BK have been implicated in the development of choroid plexus neoplasms. [17] CPTs commonly present with raised intracranial pressure symptoms. [8],[15],[18] Increased intraventricular pressure is secondary to an increase in the production of cerebrospinal fluid (CSF) by the tumor [18] and it is well documented that CPTs may produce CSF in amounts far exceeding the average of 450 mL per day that is normally observed. [15] Hydrocephalus can also be due to simple obstruction of CSF flow from large tumors [4] or impaired CSF absorption at the subarachnoid space secondary to hemorrhage or proteinaceous material from these highly vascular tumors. If undetected for long, CPTs may present with complications of chronic raised intracranial pressure-like CSF rhinorrhea and visual deterioration. Visual deterioration and opisthotonic posturing due to herniation can occur even in children with open fontanelle. [11] Macrocephaly, diencephalic seizures, gait unsteadiness, endocrine disorders are some of the other presenting features of CPT. [3] Diencephalic seizures and bobble-head doll syndrome are due to direct/indirect compression on the thalamus. Tumors that have a pedicular attachment in the ventricle may move within the ventricle, giving rise to acute gravity-dependent intermittent ventricular obstruction, and have been associated with the bobble-head doll syndrome in some cases. [4],[19] Other clinical findings include focal neurologic deficits, seizures, coma, and even psychosis. [20] Cerebellopontine angle lesions present with cranial nerve deficits and fourth ventricle lesions with cerebellar signs. Our findings are similar to the general observation but we had an unusual case of a 2-year-old girl who presented with polyphagia and obesity due to a large intraventricular CPT with third ventricular extension. Unfortunately, her symptoms failed to improve in spite of total tumor excision. CPTs are generally seen as isodense to hyperdense intraventricular masses without brain invasion on nonenhanced computerized tomography (CT) images and show intense enhancement on contrast-enhanced images. Hydrocephalus is very common as was seen in 80% of our patients. Engulfment rather than invasion of the choroid plexus glomus has been described as a means to differentiate CPTs from other trigonal lesions. [21] Calcification is generally noted in 4-10% of CPTs on plain skull radiographs and in 24% on CT images. We observed a higher incidence with nearly 50% having evidence of calcification on plain CT scan, a finding similar to Behari et al. [11] Extension from one ventricle to another or into the cerebellopontine angle is a characteristic feature of CPTs as was seen in 3 of our cases. On ultrasonography, CPPs appear as lobulated, uniformly echogenic masses. On MR imaging, CPPs appear as isointense to hypointense intraventricular masses on short transmitted reference (TR) pulse sequences and appear as variable signal intensity masses on long TR pulse sequences. Flow voids, consistent with flowing blood, are common. Distinguishing papillomas from carcinomas based on MRI scans is difficult-papillomas tend to appear as lobulated, homogeneous, enhancing masses, whereas carcinomas appear more heterogeneous because of areas of necrosis, calcification, hemorrhage, and surrounding edema. The findings of extraventricular extension of a CPT into the brain parenchyma favor the imaging diagnosis of a CPC. [4] The degree of hydrocephalus in CPCs has also been noted to be less than that seen in CPPs. [4] On positron emission tomography scan, an increased metabolic activity consistent with increased glycolysis has been seen in a CPC. [22] Screening of the spine for possible metastasis should be part of routine preoperative and postoperative investigation protocol. Enlargement of a choroidal artery is a common feature on angiographic images. [15] CPTs located in the lateral ventricle are typically supplied by the anterior choroidal artery, the lateral posterior choroidal artery, and medial posterior choroidal artery. [4] Fourth ventricular CPTs are usually supplied by choroidal branches of the posterior inferior cerebellar artery. [4],[20] High-quality MR angiography obviates the need for angiography in most cases. [4] Diffuse choroid plexus hyperplasia (CHP), an entity first described by Davis in 1924, presents with communicating hydrocephalus due to excessive production of CSF and is a close differential diagnosis for CPP. The diagnosis of CHP, is usually by exclusion of bilateral CPPs and on histologic evidence of normal choroidal morphology. CHP is usually seen in infants and arises symmetrically within both the lateral ventricles. A papilloma implies a discrete mass, whereas a CHP is defined as diffuse enlargement of the entire choroid plexus throughout the choroidal fissure, and histologically, hyperplasia with an increased number of normal-sized cells is observed). CHPs produce a large volume of CSF, reaching 4 or 5 times that of normal production much higher than that seen with CPP and the resultant hydrocephalus is also more severe. Excessive production of CSF in CHP is not resolved until the hypertrophied tissues are excised or destroyed. On MRI, the enhancing bulky appearance of the choroid plexus is the characteristic appearance of a CHP. Recent immunohistochemical staining studies, however, suggest that CPH may be a low-grade variant in the spectrum, including CPP and CPC. Surgical resection is considered to be the most effective treatment for both CPPs and CPCs, with gross total removal being the ultimate goal. [4],[7],[10],[11],[15],[23] Since the first surgical attempt at removal of a CPT in 1902, operative outcomes have markedly improved thanks to the improvement in surgical techniques. [20] We attempt to first identify and coagulate the vascular pedicle of the tumor and whenever it is not possible, bulk tumor removal is accomplished as rapidly as possible, to bring the bleeding under control. Coagulation of the tumor surface and excision in toto is preferable, but not always possible. Piecemeal resection carries the risk of life-threatening blood loss, especially in children with low blood volumes. A prompt replacement of volume and adequate suction to keep the field of resection relatively clear are mandatory adjuncts. Postoperative complications, however, are common in the form of pneumocephalus and brain shifts as highlighted by Rajkumar et al. [3] Steinbook et al. [6] too have stressed the importance of subdural collection and that preoperative VP shunt does not reduce the risk as it is associated with the ventriculo-subdural fistula and is related to the cortical thickness. Some authors have advocated shunts, [24],[25] while some have advised irrigation of the track with saline prior to closure. [3] Pneumocephalus was a major postoperative problem for us with 10 of our patients developing pneumocephalus although none required drainage. We, however, had a low incidence (8/25) of major subdural hygromas, 2 of whom required subduroperitoneal shunts. Persistent hydrocephalus is another complication that results from repeated hemorrhages resulting in basal arachnoid scarring, prolonged external ventricular drainage, increased ependymal secreting surface, and probably ependymitis. Twenty patients in our series had evidence of preoperative hydrocephalus of which 2 required VP shunts prior to definitive surgery and 4 required VP shunts after definitive surgery. Patients who did not have preoperative hydrocephalus did not develop hydrocephalus postoperatively. On gross examination, CPCs typically have a friable papillary or "cauliflower-like" appearance. [1],[26] CPTs are categorized on the basis of histologic criteria as CPP (World Health Organization [WHO] grade I), CPC (WHO grade III), and an in-between form termed atypical plexus papilloma. Two thirds of CPTs occur as the benign, slowly growing CPP, with a favorable overall prognosis, [1] whereas the rest (29-33%) manifest as a much more biologically aggressive CPC, which is far more common in children than adults. The distinction between CPP and CPC can be difficult and their histologic features may have no correlation with biological behavior. [23],[26],[27] CPPs have histologic features that are very similar to those of the normal choroid plexus. The histologic criteria for malignant tumors of the choroid plexus, that is, CPCs were first developed by Lewis in the 1960s, refined by Russell and Rubinstein 2 decades later, and most recently recodified by the WHO. The established criteria are as follows: (1) obvious invasion of adjacent neural tissue with the infiltrating cells on a stromal base, with a diffuse and poorly defined pattern of growth; (2) loss of regular papillary architecture; and (3) evidence of cellular malignancy, characterized by increased mitotic activity, nuclear atypia, and necrosis. The term atypical CPP is used [1] occasionally for a tumor with a predominantly papilloma appearance, which may have 1 or 2 malignant features. Transformation from a CPP to a CPC has been reported in a small number of cases. [27] Regardless of various classifications, the histologic appearance may not correlate with the biologic behavior [27] and many highly anaplastic CPTs can actually be clinically benign, whereas others are histologically benign, yet invasive. Both types of CPT can disseminate throughout the neuraxis and seeding of cells occurs in both CPPs and CPCs. [9],[28] However, clinically significant seeding leading to frank metastatic spread is much more common in patients with a carcinoma [1] and the rate of dissemination of CPC within the neuraxis is about 30%. [9],[28] CPCs typically stain positive for cytokeratins, and display variable expression of vimentin, S100, transthyretin, and glial fibrillary acidic protein (GFAP). Positivity for S100 and transthyretin is typically less than that seen in CPP. CPCs stain positive for GFAP in approximately 20% of tumors, and they can express carcinoembryonic antigen. CPCs are typically negative for epithelial membrane antigen. Immunohistochemical assays for prealbumin and carcinoembryonic antigen expression are of significant value for the differential diagnosis of CPPs and CPCs and that high Ki-67 staining index values may serve as indicators of CPP recurrence, even if the primary lesion is benign. Coons et al. has demonstrated that a high proliferation rate, as inferred from higher S-phase fractions, is generally associated with a poorer outcome. MIB-1 labeling is an easier method for assessing choroid plexus neoplasms than is analysis of differentiation markers. The expression of CD44 effectively documents invasive potential and may predict tumor progression. Histologically, the atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system have overlapping histologic features. [16],[28] Although the hSNF5/INI1 mutation, commonly present in AT/RT, has also been found in CPC, [29],[30] more recent data have shown that immunostaining for INI1 is retained in the majority of CPCs, which may help to distinguish CPC and AT/RT. [30] The prognosis for CPP patients treated with gross total resection (GTR) is excellent, with a report of 100% survival at 5 years after surgical resection in many series including ours, and adjuvant therapy is not indicated in these patients. Radiotherapy is not necessary following GTR, and its usefulness should be reserved for recurrent disease. Unfortunately, the prognosis for patients with CPC is guarded, with an overall 5-year survival rate of 26-50% [5],[16] GTR is generally considered the most important prognostic factor [31] for CPC as was confirmed in our analysis. The 5-year survival rate of 58% after complete tumor resection (vs 20% after partial resection) was in agreement with previous reports. [16],[27],[31] The median survival for CPCs who underwent gross total excision with adjuvant therapy was 58 months and those who had a subtotal resection with adjuvant therapy was 36 months. Even after primary complete resection of CPC, there are significant relapse rates, [31] which might be due to microscopic residual tumor after macroscopically complete tumor resection. Additional surgery should be performed in subtotally resected patients when it is likely to result in a GTR with a good functional neurologic outcome. The role of chemotherapy for CPC is less clear. There are several reports of small series suggesting an effect of postoperative chemotherapy in patients with tumors that are less than completely resected, when the patient is too young to receive radiation, whereas the role of neoadjuvant treatment after complete resection and that of chemotherapy in addition to radiation is controversial. [16] The late neurologic sequelae of radiotherapy given to children younger than 3 years exclude this modality, leaving only chemotherapy as an adjuvant treatment for the majority of patients with CPC. Chemotherapy should be given to patients with less than completely resected CPC, if possible in combination with radiotherapy. Current data strongly support the use of combined radiochemotherapy in patients older than 3 years and chemotherapy alone if the patients are younger. The total amount of necessary adjuvant treatment and the order in which the modalities are to be used are still controversial. [16],[31] Conclusion CPTs are rare intracranial tumors and include a spectra ranging from choroid plexus hyperplasia to CPP to CPC. Radiologic and histologic characterization of these tumors is difficult and newer immunohistochemical and genetic studies should be done to differentiate them from each other. Total excision offers good prognosis and should be attempted for all forms of CPTs. CPPs carry a good prognosis and adjuvant therapy is not indicated even after partial excision. CPCs and atypical CPCs carry a poor prognosis, and adjuvant therapy improves survival marginally after total excision. Spinal drop metastases are common for CPC and screening of the spine for possible metastasis should be part of routine preoperative and postoperative investigation protocol. References
Copyright 2010 - Neurology India The following images related to this document are available:Photo images[ni10109t3.jpg] [ni10109t4.jpg] [ni10109t1.jpg] [ni10109f3.jpg] [ni10109f1.jpg] [ni10109t2.jpg] [ni10109f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}