 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
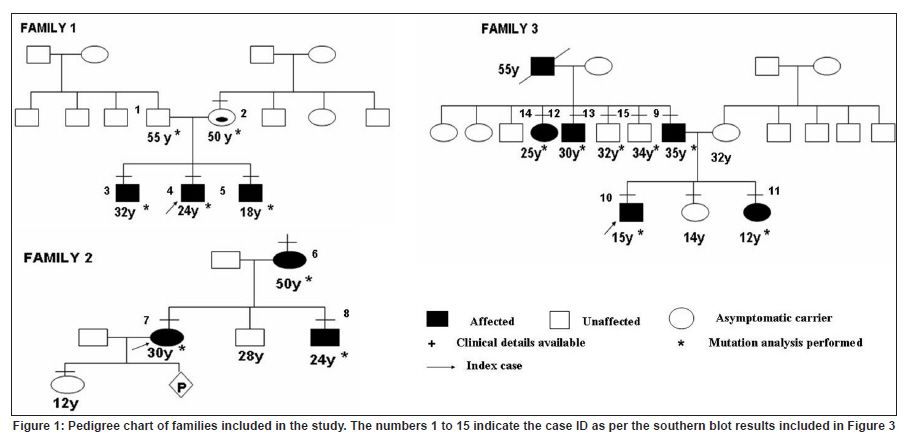
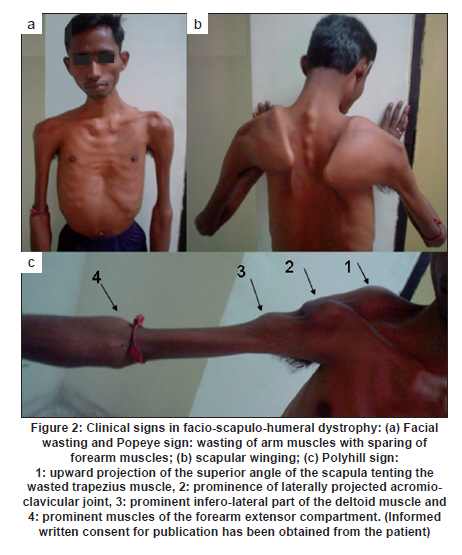
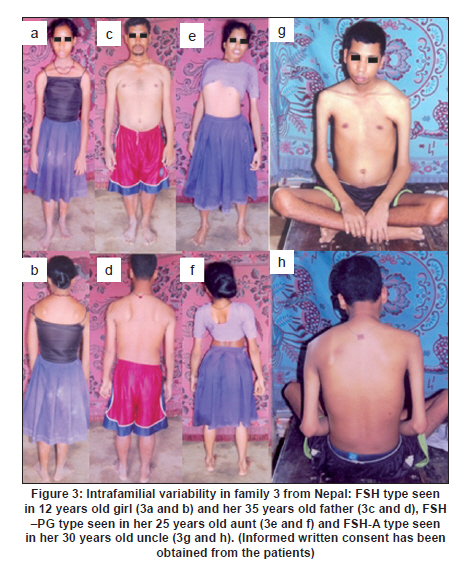
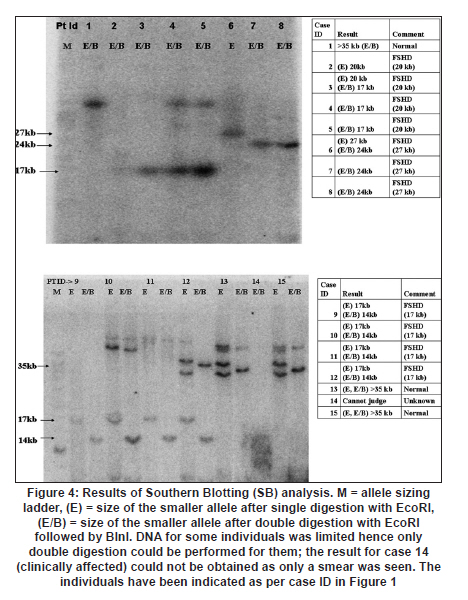
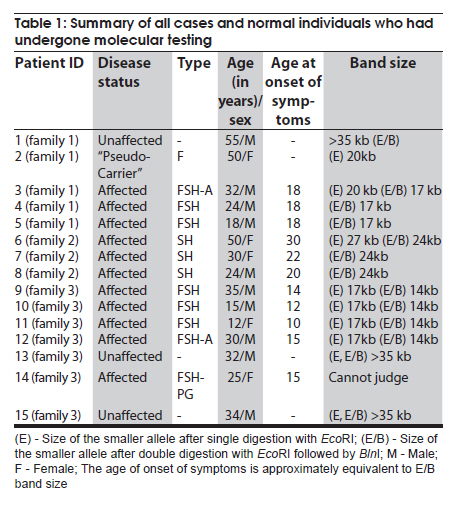
Neurology India, Vol. 58, No. 3, May-June, 2010, pp. 436-440 Brief Report Clinical profile and molecular diagnosis in patients of facioscapulohumeral dystrophy from Indian subcontinent Parag M Tamhankar, Shubha R Phadke Department of Medical Genetics, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Rae Barely Road, Lucknow - 226 014, Uttar Pradesh, India Date of Acceptance: 23-Oct-2010 Code Number: ni10110 PMID: 20644274 Abstract Facioscapulohumeral dystrophy (FSHD) is an autosomal dominant muscular dystrophy. We retrospectively studied three families (two Indian, one Nepalese) with 12 affected members (male:female-7:5). Mean age at onset of weakness was 17.63 + 5.48 years. Patients were classified according to muscle groups affected (F-face, S-scapula, H-humeral, PG-pelvic girdle, P-peroneal, A-loss of independent ambulation): FSH-A (2), four FSH (4), SH (3), FSH-PG (2) and one: F (1). Progression of weakness was classified as F>S>P>PG in eight cases, S> F>P in one, static in three. Eleven patients had electromyographic findings suggestive of myopathy and one had features of neurogenic involvement. Molecular diagnosis was done by southern blotting using probe p13E-11 after digestion of genomic DNA with EcoRI and/or EcoRI/BlnI for twelve patients and three unaffected relatives. No EcoRI fragment smaller than 35 Kb was seen in unaffected subjects. Size of EcoRI fragment varied between 17 kb to 27 kb in affected subjects and was constant for affected members of the same family. Molecular diagnosis by southern blotting has helped to provide genetic counseling for the families. Keywords: BlnI, facioscapulohumeral dystrophy, EcoRI, 4q35 Introduction Facioscapulohumeral muscle dystrophy (FSHD) is an autosomal dominant disorder which characteristically affects the facial, scapular and humeral muscles and eventually the pelvic and lower limb muscles. It accounts for only 2 to 3% of muscular dystrophy cases in India. [1] The gene for FSHD has been mapped to 4q35 locus (sub-telomeric region of long arm of chromosome 4), which contains several 3.3 kb KpnI repeats. Molecular diagnosis is done by performing conventional southern blotting (SB) with probe p13E-11 (the most telomeric probe of 4q35 region) followed by double digestion of genomic DNA by EcoRI and BlnI restriction enzymes. EcoRI fragment sizes vary between 50 kb to 300 kb in normal individuals and are lesser than 35 kb in 95% of FSHD patients due to decreased number (deletion) of KpnI repeat. [2] The published data on this disorder in the Indian subcontinent is scarce. We present the clinical profile and mutation analysis in patients with FSHD from India and Nepal. Materials and Methods Clinical profiling: Case records of patients with FSHD diagnosed between the years 2004 to 2008 were reviewed retrospectively. Clinical history, details of neurological examination and investigations were noted. Pop-eye sign, Pradhan's poly-hill sign and Beevor sign were elicited as previously described. [3],[4] Cases were classified according to the muscle groups presently affected (F-face, S-scapula, H-humeral, PG-pelvic girdle, L-leg, A-loss of independent ambulation). Progression of weakness was classified as facio-scapulo/brachio-peroneo-pelvic (FSPPG) (order of weakness: face?upper limb/scapula?lower limb), or scapulo/brachio-facio-peroneal (SFP) (scapula/upper limb?face?lower limb) or static. Molecular diagnosis: The study protocol was approved by the institute's ethics committee. Informed written consent for research and publication was taken from patients and their families. Genomic DNA was isolated from peripheral blood (10 ml) by standard phenol-chloroform method. Genomic DNA was initially digested with EcoRI for twelve hours at 37o C as suggested by the manufacturer's protocol. The DNA was separated on a 0.3% HGT Agarose gel for 72 hours at 15 mA (0.5 V/cm) and was transferred to Hybond N+ (Amersham). The probe p13E-11(D4F104S1) (0.8 kb insert in pBS can be isolated by SacI/EcoRI double digestion) was labeled with 32P-dCTP using a Random Primed DNA labeling kit (Takara). Hybridisation was carried out at 65C. The filter was washed in a stringency of 2 Χ SSC/0.1% SDS for 20 minutes X 2 at 65C, followed by autoradiography for two hours using BAS 2500 image analyzer (Fuji) and Konica AX film with an intensifying screen for three days. Genomic DNA of individuals having EcoRI fragment size less than 35 kb was double digested with EcoRI and BlnI together and analyzed as for EcoRI. [2] Results Three families with 12 affected members were included (male: female ratio: 7:5) [Figure - 1]. In family one, the mother (patient 2) was a "pseudo-carrier" (asymptomatic but examination revealed mild facial wasting and weakness and she carried the mutation). She refused to be further investigated. The mean age at presentation was 29.08 ± 12.30 years. The mean age at the onset of weakness (n=11, one patient was asymptomatic) was 17.63 ± 5.48 years (range 10-30 years). Symptoms included muscular pain (9/12) and weakness (11/12). The frequency of various clinical symptoms/signs were facial weakness (9/12); scapular winging (11/12); polyhill sign (10/12); pop-eye sign (9/12); pelvic girdle weakness (4/12); peroneal muscle weakness (4/12); and Beevor sign (11/12) (see [Figure - 2] and [Figure - 3] for clinical signs). Asymmetric limb involvement was noted in 8/12 patients and asymmetric facial involvement was noted in 4/9 patients. Two patients had FSH-A with onset of weakness at ages 15 and 20 years and loss of ambulation at ages 25 years and 30 years respectively. Other patients were: FSH (4), SH (3), FSH-PG (2) and F (pseudo- carrier, 1). The progression of weakness was FSPPG in eight patients, SFP in one and static in three. None of the affected patient had ophthalmoplegia, respiratory or pharyngeal muscle weakness, fundus changes, hearing loss, psychiatric symptoms or epilepsy. The mean serum creatine phosphokinase levels were 502.16 ± 331.01 IU/l (n = 6; range: 40 to 1050). Electromyography (EMG) showed myopathic findings in the majority (5/6, 83.33%) and neurogenic findings (spontaneous activity and increased positive sharp waves) in the remaining patient (1/6, 16.67%). Muscle biopsy was not performed in any of the patient. Electrocardiogram did not show any abnormalities. Inheritance pattern observed in all the families was autosomal dominant. Mutation analysis was possible in 12 affected subjects and three unaffected family members. All the affected subjects were heterozygotes for an EcoRI fragment smaller than 35 kb size and a normal sized allele (EcoRI fragment more than 35 kb). The unaffected family members tested did not demonstrate EcoRI fragment smaller than 35 Kb. The allele size remained constant for affected subjects in the same family [Figure - 4]. However, intrafamilial variability in clinical presentation and progression was seen in family number 1 and 3 [Figure - 1] and [Figure - 3] and [Table - 1]. One of the patients (family 2) had presented for prenatal diagnosis and counseling at 24 weeks of gestation. The 30 year-old affected patient was counseled that the risk of recurrence in her baby was 50% as the inheritance pattern in her family was autosomal dominant. Prenatal diagnosis could not be offered owing to lack of mutation facilities at our laboratory then. The patient opted for continuation of pregnancy. All affected subjects were advised physiotherapy and supportive care. Discussion In FSHD penetrance of the disease is high and majority of the affected cases (95%) have some weakness by 20 years of age. [4] Late presentation of patients to our tertiary care center could be due to delayed referral and lack of awareness of the disease amongst primary care physicians. The typical scattered and asymmetric pattern of weakness and muscular pain and resultant clinical signs observed in our patients was attributed to the muscle inflammation. [5] The frequencies of the typical signs and symptoms seen in this series are similar to that reported in literature [4] The typical progression of weakness (FSPP) ("descending" type) is early facial and scapular involvement followed by upper arm involvement with eventual spread to abdomen and pelvic girdle. [4] The "ascending" type (SFP) was uncommon and occurred when the facial muscles were involved later than the scapulo-humeral. This type is likely to be confused with limb girdle muscle dystrophy or late onset spino-muscular atrophy. Patients with FSHD have normal life expectancy and it is because of the slow progression of the disease, lack of respiratory muscle involvement (<1%) and low incidence of cardiac arrhythmias (5%). [4] Atypical cases with vision and hearing loss, epilepsy, mental retardation, ophthalmoplegia, unilateral brachial weakness, childhood and infantile forms are a minority and were not seen in our study. [4] Diagnosis of extra retinal vasculopathy and sensorineural deafness may need fluoroscein angiography and pure-tone audiometry respectively and were not performed routinely in our patients. [4] Serum creatine kinase (CK) levels are typically mildly to moderately elevated (<1500 IU/l). [6] EMG shows myopathy features in most cases but neurogenic features such as spontaneous muscle activity, large muscle compound action potentials as seen in one of our patients can occur and have been attributed to muscle hypertrophy rather than denervation and re-innervation phenomenon. [7] Muscle biopsy may show signs of inflammation in addition to myopathic features; however, it is not essential for the diagnosis. In patients with atypical clinical and/or molecular findings muscle immuno-histochemistry may be needed to differentiate from sarcoglycanopathies. [4] Molecular diagnosis by southern blotting technique shows 95% sensitivity and 95% specificity for diagnosis of FSHD. [6] The remaining is labeled as non-4q35 linked FSHD. The latter was not observed and all patients in our series were linked to 4q35. The number of D4Z4 repeats could be calculated as EcoRI fragment size minus 5 Kb divided by 3.3. The allele sizes detected viz. 27 kb, 20 kb and 17 kb thus correspond to approximately 7, 5 and 4 D4Z4 repeats respectively. There is only one previously published study from India (Chhattisgarh state) that has documented that FSHD cases in India are also linked to 4q35. The smaller EcoRI fragment was 26 kb in three patients from a single family. [8] The range of size of pathogenic FSHD alleles may vary between different countries. In Japan, EcoRI alleles are commonly between 10-25 Kb and in Caucasians between 10- 40 kb. Our study had alleles ranging from 17 kb to 27 kb. Long range polymerase chain reaction (PCR), the newer rapid method of molecular diagnosis of FSH amplifies pathogenic alleles above 25 kb (approximately 5 D4Z4 repeats) with difficulty. [9] Use of Long PCR may thus result in diagnostic difficulty in at least some of our families. Knowledge of allele size also helps in predicting onset, severity and progression of FSHD. In our study, milder forms (SH) were associated with 27 kb allele size and more severe forms- FSH, FSH-PG and FSH-A were associated with allele sizes (viz. 17 kb and 20 kb). Allele sizes between 35 kb to 50 kb, usually labeled as inconclusive, may occur in patients with typical FSHD. Normal individuals have allele sizes above 50 kb. Fragment sizes above 35 kb cannot be resolved by SB but can be resolved by pulse field gel electrophoresis. We did not come across allele sizes in this range. The meiotic stability of the D4Z4 allele is demonstrated by constant allele size in affected subjects in a family through successive generations. Anticipation was observed in family numbers 1 and 2 wherein the mother had transmitted the disease to her children and not in family 3 wherein the father had transmitted the disease. This gender-specific anticipation in FSHD is known but the molecular basis remains largely unexplained. This is in contrast to repeat expansion disorders such as Huntington disease, Fragile X etc which show anticipation due to repeat expansion in successive generations due to germ cell specific meiotic repeat size expansion. [10] Southern blotting is laborious, time consuming and the D4Z4 allele size is roughly estimated from size of the EcoRI fragment. Long PCR accurately identifies 90% cases of FSH by using primers specific to the 4q35 locus. It is faster, requires 1/100 th of genomic DNA required in southern blotting removes need for enzyme digestion, does not involve use of radioactive isotopes and is 1/30 to 1/40 times less costly. [11] This method could not be performed as DNA was limited. We also could not perform "qA/qB allele" typing as DNA was limited. This typing refers to biallelic variation named qA and qB identified at 4q telomeric region distal to D4Z4. The smaller alleles linked to FSHD are exclusively of qA type. [12] However, unaffected individuals having small alleles (<35 kb) that are linked to qB alleles would be very uncommon. Acknowledgment We acknowledge the support of the patients and their families for this research. We also thank the Indian Council of Medical Research for funding our DNA banking facility. We acknowledge Dr. Kanako Goto and Dr. Yukiko K. Hayashi for performing the southern blotting analysis at the National Institute of Neuroscience, National Center of Neurology and Psychiatry, Kodaira, Tokyo, Japan. References
Copyright 2010 - Neurology India The following images related to this document are available:Photo images[ni10110t1.jpg] [ni10110f3.jpg] [ni10110f2.jpg] [ni10110f4.jpg] [ni10110f1.jpg] |
| |||||||||


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}