 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
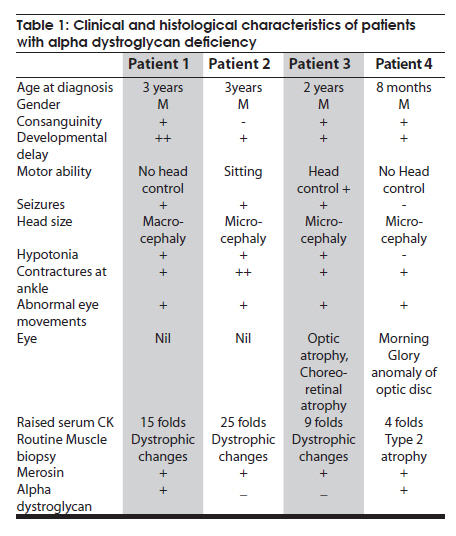
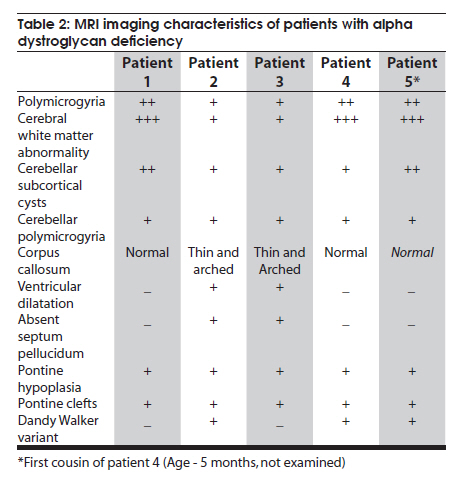
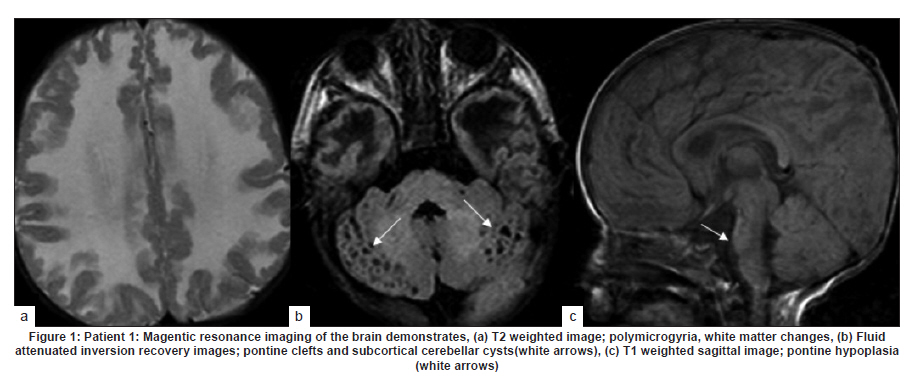
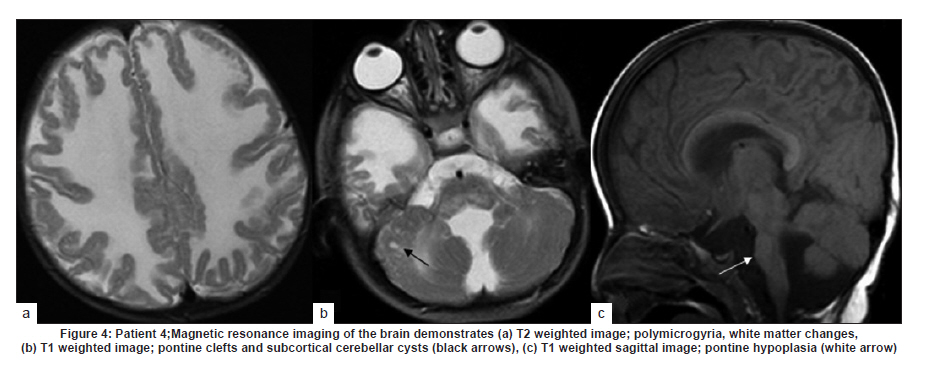
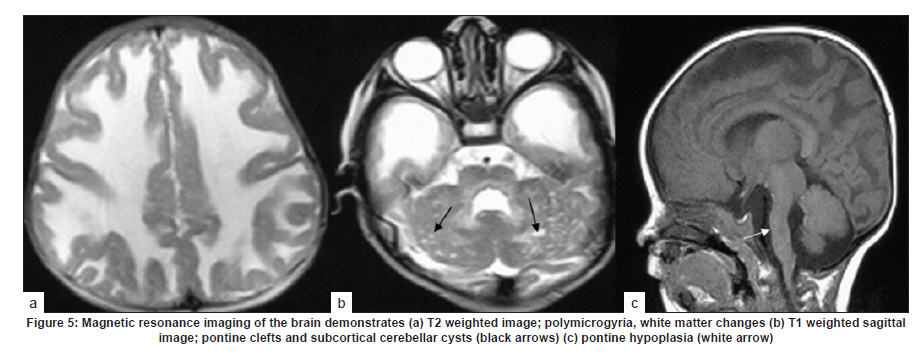
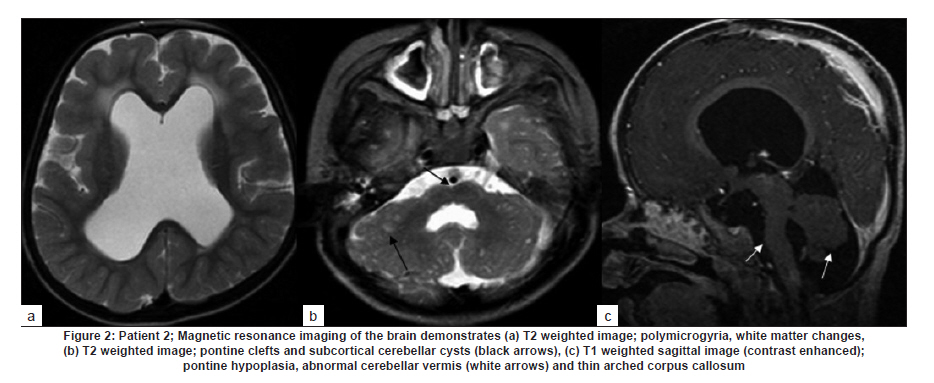
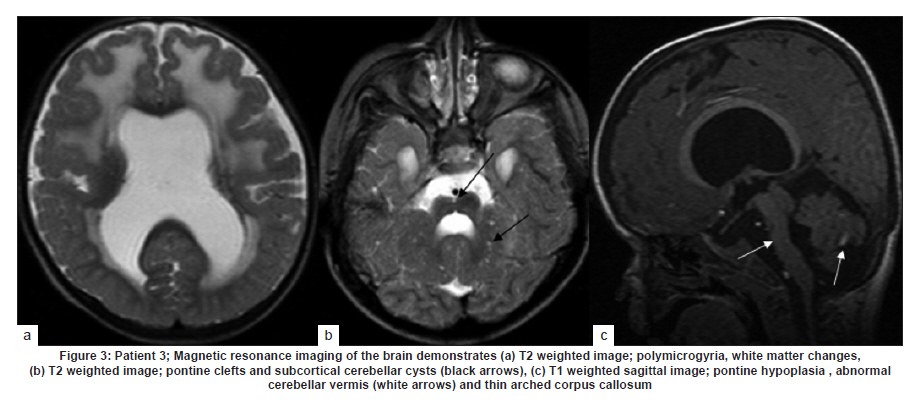
Neurology India, Vol. 58, No. 3, May-June, 2010, pp. 460-465 Case Report Pattern recognition on brain magnetic resonance imaging in alpha dystroglycanopathies Parayil S Bindu1, Narayanappa Gayathri2, Rose D Bharath3, Anita Mahadevan2, Sanjib Sinha1, AB Taly1 1 Department of Neurology, National Institute of Mental Health And Neurosciences (NIMHANS), Bangalore, India Date of Acceptance: 08-Jun-2010 Code Number: ni10117 PMID: 20644281 Abstract Alpha dystroglycanopathies are heterogeneous group of disorders both phenotypically and genetically. A subgroup of these patients has characteristic brain imaging findings. Four patients with typical imaging findings of alpha dystroglycanopathy are reported. Phenotypic features included: global developmental delay, contractures, hypotonia and oculomotor abnormalities in all. Other manifestations were consanguinity (3), seizures (3), macrocephaly (1), microcephaly (3), retinal changes (2) and hypogenitalism (2). Magnetic resonance imaging (MRI) of the brain revealed polymicrogyria, white matter changes, pontine hypoplasia, and subcortical cerebellar cysts in all the patients, ventriculomegaly, callosal abnormalities, and absent septum pellucidum in two and Dandy -Walker variant malformation in three. Magnetic resonace imaging of the first cousin of one the patient had the same characteristic imaging features. Brain imaging findings were almost identical despite heterogeneity in clinical presentation and histopathological features. Pattern recognition of MR imaging features may serve as a clue to the diagnosis of alpha dystroglycanopathy. Keywords: Alpha dystroglycanopathy, brain malformations, cerebellar cysts, congenital muscular dystrophy, polymicrogyria, pontine hypoplasi Introduction Identification of congenital muscular dystrophy (CMD) syndromes with deficient glycosylation of alpha dystroglycan is an important development in the field of congenital muscular dystrophy. [1],[2] Alpha dystroglycan, an integral component of dystrophin glycoprotein complex, is also expressed in the developing central nervous system, retina, and cochlea and plays an integral role in neuronal migration, organization of synapses and assembly of basement membrane. [3] Alpha-dystroglycanopathies are clinically heterogeneous, involve multiple organ systems and result in severe brain malformation and developmental delay and are caused by genetic defects that disrupt posttranslational modification of alpha dystroglycan and other currently unknown proteins. [4] Previously these patients were classified as CMD with brain malformations such as Fukuyama disease, Walker Warburg syndrome and Muscle Eye brain disease. Attempts have been made to classify them based on magnetic resonance imaging (MRI) features. [5],[6] In this report we describe four patients with characteristic MRI findings of alpha dystroglycanopathy and highlight the role of neuroimaging in the diagnosis. Patients and Methods The diagnosis of alpha dystroglycanopathy in these four patients was based on the combination of clinical and histological features of CMD, severe mental retardation and evidence of brain malformations on MR imaging. Skeletal muscle biopsies (vastus lateralis -3, biceps- 1) were processed for routine histology and a battery of enzyme histochemical stains (succinic dehydrogenase (SDH), nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR), Modified Gomori trichrome (MGT), adenosine triphosphatase (ATPase) at pH 9.4 and 4.6). Immunohistochemistry by indirect immunoperoxidase technique was carried out using monoclonal antibodies (dystrophin 1, 2, 3, sarcoglycan (α, β, γ), dysferlin , alpha 2 laminin(Merosin), β dystroglycan (Novocastra laboratories,Newcastle upon Tyne) and Alpha dystroglycan (VIA4-monoclonal antibodies, Millipore Corporation, USA). Brain MRI sequences included: T1-weighted axial and sagittal images (TR/TE =600/14 ms) and T2 weighted axial and coronal images (TR/TE = 5000/90 ms), fluid attenuated inversion recovery sequences (TR/TE= 9000/150 ms) in all the patients. Brain MRI observations in the first cousin of patient 4 who had identical clinical history (not examined) was also included for analysis. Results The details of the clinical, laboratory and histopathological and MR imaging features of all the four patients are provided in [Table - 1] and [Table - 2]. Clinical features All the four patients had mental retardation, weakness, hypotonia, contractures and oculomotor abnormalities. Other manifestations were consanguinity (3), seizures (3), macrocephaly (1), microcephaly (3), retinal changes (2) and hypogenitalism (2). Presenting complaints in the four patients were different and included progressive macrocephaly (Patient 1), behavioral abnormalities like head banging, frequent tantrums (Patient 2), seizures (Patient 3) and isolated developmental delay (Patient 4). There was optic atrophy and choreo-retinal atrophy in patient 2 and morning glory anomaly of the optic disc in patient 4. Histological features Muscle biopsies showed dystrophic features with rounding, myofibre size variation myonecrosis, regeneration and endomyseal fibrosis on routine histology in three patients while type 2 atrophy was noted in one. Immuno labeling to merosin was preserved in all while staining to a-dystroglycan showed total absence/deficiency in two patients and preserved labeling in two. Imaging features Brain MRI in all the patients showed a combination of cortical malformation, white matter signal intensity changes and posterior fossa malformations [Table - 2]. In three patients imaged in infancy, cortical malformations were characterized by multiple small gyri and irregular bumpy grey white junction [Figure - 1]a, [Figure - 4]a and [Figure - 5]a. In patients aged above one year, the cortical abnormalities showed a thicker cortex with superimposed irregularities [Figure - 2]a and [Figure - 3]a. Gyral abnormalities were more prominent in the frontotemporal area. Cerebral hemispheric white matter abnormalities were present in all. In three patients below one year of age, white matter signal intensity changes were extensive and involved all the lobes diffusely [Figure - 1]a, [Figure - 4]a and [Figure - 5]a. On T1 weighted image, myelination was present only in posterior limb of the internal capsule, splenium and optic tract and some parts of the brain stem. In the two patients imaged after infancy signal intensity changes in the white matter were less extensive. In patient 2, the signal intensity changes were present in the periventricular and deep white matter in the frontal region and involved only periventricular region in the parietotemporal lobes [Figure - 2]a. In patient 3, the changes were prominent in the periventricular, deep white matter and subcortical U fibers of the frontal lobe and limited to periventricular white matter in parietal and temporal regions [Figure - 3]a. Posterior fossa abnormalities were present in all. There was flattened ventral surface of the pons indicating pontine hypoplasia [Figure - 1]c, [Figure - 2]c, [Figure - 3]c, [Figure - 4]c and [Figure - 5]c and ventral pontine clefts [Figure - 1]b, [Figure - 2]b and [Figure - 3]b. There was hypoplasia of the cerebellar vermis with rotated vermis giving the appearance of Dandy-Walker variant type of configuration in three patients [Figure - 2]c, [Figure - 3]c and [Figure - 4]c. Cerebellar subcortical cysts were present in all and were most prominent in patient 1 [Figure - 1]b, [Figure - 2]b, [Figure - 3]b, [Figure - 4]b and [Figure - 5]b. Discussion Alpha dystroglycanopathies consists of a spectrum of disorders: Fukuyama congenital muscular dystrophy, Walker Warburg syndrome and Muscle Eye brain disease; Merosin deficient CMD 1C and 1D (MDC1C, MDC1D); limb girdle muscular dystrophy 2I. The clinical features, pathological and imaging findings in our patients conform to CMD with brain malformations: Muscle eye brain disease phenotype-2 (Patient 3, 4) and Fukuyama phenotype-2 (Patient 1, 2). It is noteworthy that these patients also had some overlapping features of Walker Warburg phenotype such as macrocephaly and hypogenitalism. [7] Patient 4 had "morning glory" anomaly of the optic disc, an unusual ophthalmological finding. It is intriguing that despite involvement of muscle, eye and brain in patient 4 and muscle and brain involvement in patient 1, immunohistochemistry showed preserved staining for alpha dystroglycan; neverthless, these two patients had the MR imaging findings typical of alpha dystroglycanopathies. In our study, only the VI 4A- 1 clone of the antibody directed against a glycosylated epitope of the alpha dystroglycan was used. Use of additional clones of antibodies like IIH6 in combination with antibodies directed against the core protein or quantification by Western blot is recommended to confirm alpha dystroglycan deficiency in those patients where immunostaining against one epitope fails. The differential staining of the tissues with different clones of antibodies as well as variation in staining with different batches of antibodies have been previously reported. [8],[9] Genetic testing is an alternative to elucidate the exact subtype and nature of the disease. However, absence of demonstrable genetic mutations would not exclude this group since a large number of patients with the same phenotype did not have any known genetic mutations in a recent study. [10] Both western blotting and genetic confirmation could not be done in our patients because of non availability. The constellation of the MR imaging findings in these patients viz. migration abnormalities, white matter changes, cerebellar cysts and pontine hypoplasia is striking. Alpha dystroglycan is expressed in basement membrane. Abnormal alpha glycosylation results in discontinuity of pial-glial limitans allowing migration of neurons and glia beyond the pial basement membrane. [11] In addition to the disrupted radial migration defect, morphological studies of the brainstem in Fukuyama congenital muscular dystrophy indicate perturbed tangential migration. [12] The recently described ventral pontine clefts, [13] seen in our patients is produced by defects in tangential migration, that cause absence of ventral transverse pontine fibers at the decussation of middle cerebellar peduncle. [14] Cerebellar cysts have been consistently reported in dystroglycanopathies. Cerebellum is the most vulnerable structure affected in patients with FKRP- related conditions. [15] It may be an isolated MRI finding in patients with MDC1C especially in those with mild central nervous system involvement. [15] Neuropathological studies have demonstrated that cerebellar cysts occur within or near the polymicrogyric cortex and evolve from invagination /entrapment of the subarachnoid spaces within the malformed folia. [16] White matter changes in CMD are due to transient dysmyelination and represent a common phenomenon in dystroglycanopathies. [15] Extensive white matter changes, observed in early infancy, may resolve with increasing age and remain confined to periventricular white matter suggesting that delayed myelination could be the underlying basis for these changes. In pathological studies, the white matter abnormalities correspond to myelin pallor and gliosis. [17],[18] There are only a few studies, which describe the imaging findings in CMD with brain malformations. The initial studies focused on classifying these disorders based on the MRI findings [5],[6],[16] while recent reports correlate imaging changes with genotyping. [13],[15],[19] In a recent study, brain involvement in 26 patients with dystroglycanopathies ranged from complete lissencephaly to isolated cerebellar involvement. [1] No pattern of abnormality unequivocally allowed identification of the primary gene defect. Since genetic study was not done in our patients such a comparison is not possible. The imaging changes reported are highly evocative of a dystroglycanopathy. [13] The differential diagnosis for such constellation of findings viz. cortical malformations and white matter changes include peroxisomal biogenesis disorder, and congenital cytomegalo virus infection. [20],[21] Presence of germinolytic cysts or atrophy may distinguish peroxisomal biogenesis disorder from CMD, but the distinction can at times be difficult. Brainstem malformations seen in dystroglycanopathies are not reported in latter conditions. Subtyping of CMD is important for directing genetic analyses. Conventional histology of the muscle tissue does not allow differentiation among the various subtypes. Similarly the clinical features are diverse and differentiation may not be possible in the absence of genetic testing. Our study indicates that in a subgroup of patients with CMD, brain imaging may serve as surrogate marker for alpha dystroglycanopathy. References
Copyright 2010 - Neurology India The following images related to this document are available:Photo images[ni10117f2.jpg] [ni10117f5.jpg] [ni10117t1.jpg] [ni10117f1.jpg] [ni10117f3.jpg] [ni10117t2.jpg] [ni10117f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}