 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
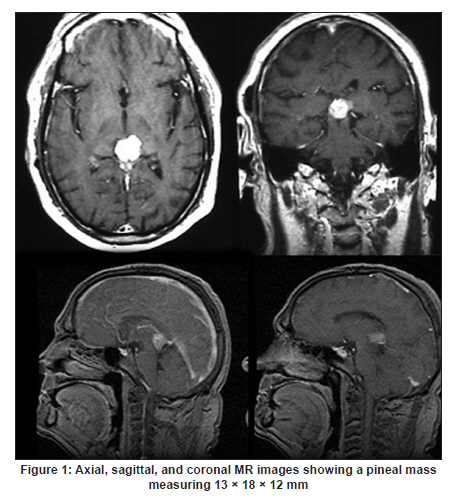
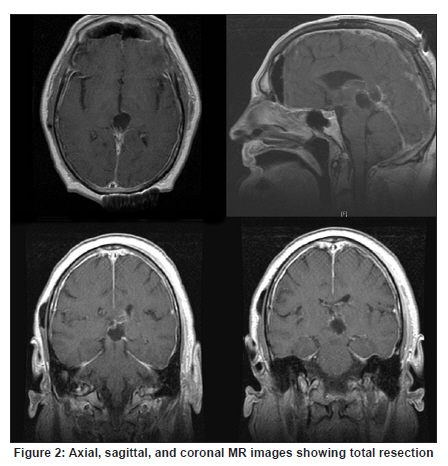
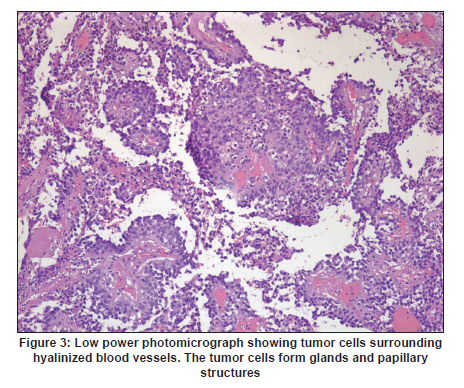
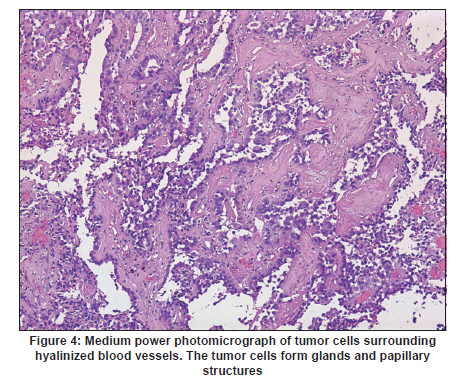
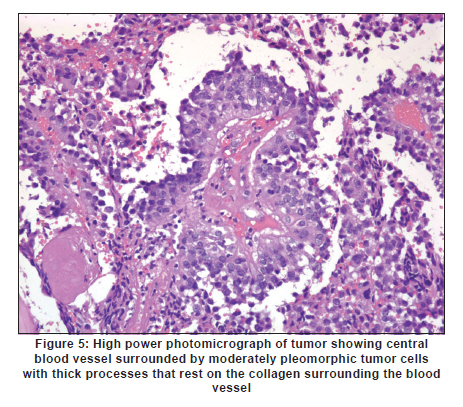
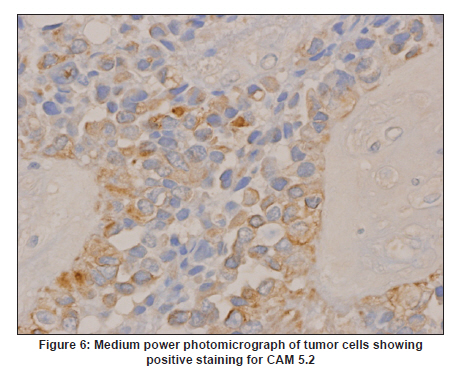
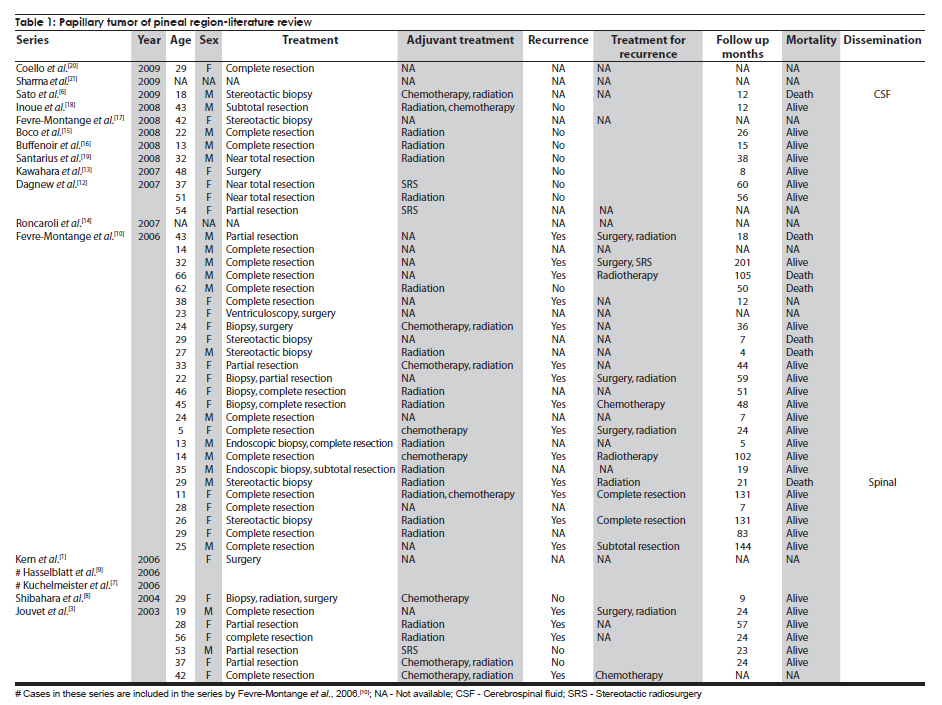
Neurology India, Vol. 58, No. 3, May-June, 2010, pp. 471-476 Case Report Papillary tumor of pineal region: Prolonged control rate after gamma knife radiosurgery - A case report and review of literature Raul Cardenas1, Vijayakumar Javalkar1, Justin Haydel1, Rishi Wadhwa1, Marjorie Fowler2, Bernd Scheithauer3, Anil Nanda1 1 Department of Neurosurgery, Louisiana State University Health Sciences Center, Shreveport, USA Date of Acceptance: 08-Jun-2010 Code Number: ni10120 PMID: 20644284 Abstract Papillary tumors of the pineal region (PTPR) are very rare. We describe the first report of a PTPR empirically managed with gamma knife radiosurgery. The patient was initially shunted and referred for empirical gamma knife radiosurgery. After initially showing some improvement, he had recurrence of tumor after 7 years. For recurrence he underwent a gross total resection and the biopsy established the diagnosis of PTPR. Further research needs to be done as to the efficacy of gamma knife surgery for PTPR. In addition, the role of stereotactic biopsy for eligible patients should be considered as the initial step to direct the treatment of choice. Keywords: Gamma knife radiosurgery, papillary tumor of pineal region, recurrence Introduction Papillary tumors of the central nervous system are rare and have been documented in the pineal region on few occasions. Tumors originating from the pineal region include the following: pineal parenchymal tumors (pineoblastoma, pineal parenchymal tumor of intermediate differentiation, and pineocytoma), astrocytoma (most commonly pilocytic astrocytoma), and germ cell tumors. [1],[2] Papillary configurations are manifested in some of these tumors, such as papillary pineal parenchymal tumor, papillary ependymoma, and choroid plexus papilloma. The first 6 cases of papillary tumors of the pineal gland (PTPR) were described by, Jouvet et al., [3] in 2003. Prior to this, only 2 reports of tumors with papillary features in the pineal region had been reported. [4],[5] In this report, we describe the first case of a primary PTPR recurring 7 years after initial empirical treatment with gamma knife surgery. Case Report A 47-year-old Caucasian man presented with a 6-month history of increasing headaches, diplopia, dizziness, and drowsiness in 2005. On neurologic examination, he did not have any focal deficits. His past history revealed that he had presented with acute hydrocephalus to an outside facility in 1998. His workup there revealed a pineal mass. For this, the patient underwent a ventriculoperitoneal shunt and was referred to gamma knife radiosurgery without tissue diagnosis at an outside facility. His initial magnetic resonance imaging (MRI) study revealed a primary pineal tumor measuring 20 Χ 21 Χ 17 mm with obstructive hydrocephalus. Following radiosurgery, the tumor reduced in size to 7 mm. His current MRI scans revealed a pineal mass measuring 13 Χ 18 Χ 12 mm [Figure - 1]. Because of the symptomatic recurrence, surgical excision was offered. A gross total resection was achieved by occipital interhemispheric approach and this was confirmed with MRI scan [Figure - 2]. The postoperative course was uneventful. In 2008, the patient underwent a shunt revision with the replacement of his original medium pressure valve with a programmable valve because of the features of over drainage. At last follow-up, 42 months after the surgical excision at our center, the patient was alive without any recurrence of the tumor. Histology and immunohistochemical findings Microscopically, the tumor sections revealed a vascular neoplasm composed of sheets of moderately pleomorphic tumor cells frequently forming rosettes around the blood vessels. The rosetted cells had thick processes resting on collagen surrounding the vessels. Many of the blood vessels within the tumor were hyalanized and masses of fibrinoid material were present within the tumor [Figure - 3], [Figure - 4], [Figure - 5]. Immunohistochemically the tumor was positive with stains against S-100, CAM 5.2 [Figure - 6], and prealbumin. The stains against alpha-fetoprotein, CD-10, synaptophysin, and placental alkaline phosphatase were negative. In addition, the tumor cells tested focally positive for glial fibrillary acid protein (GFAP) [Figure - 7]. This staining pattern supports the diagnosis of a PTPR. Discussion Recently the PTPR is recognized as a distinct entity in the 2007 WHO classification of brain tumors. [6] These tumors were tested immunohistochemically with a profile similar to that of a choroid plexus tumor; however, morphologically the tumors appeared to be less differentiated than a choroid plexus papilloma and more differentiated than a choroid plexus carcinoma. From the earlier case reports, the behavior of the tumors was not entirely clear, but it appeared to be very similar to ependymal and choroid plexus tumors, which have a tendency for local recurrence. [3],[4],[5] In the past, PTPR have been misdiagnosed as either ependymomas or choroid plexus tumors. [3],[7],[8] The histopathology of the papillary tumors resembles those of ependymomas and choroid plexus tumors. [9] According to Hasselblatt et al.,[9] the majority of the PTPR can be distinguished by the absence of epithelial membrane antigen staining, membranous Kir7.1 and cytoplasmic staniocalcin-1 staining, and the presence of MAP-2 immunoreactivity. After the first publication in 2003, Jouvet et al.[3] published the largest series of PTPR, [10] including 31 cases from multiple centers. The majority of these cases (68%) had a gross total resection; nevertheless, the 5-year estimates for overall survival and progression-free survival were 73% and 27%, respectively. [10] In 2006, the same group had published the details about immunohistochemical profile and chromosomal imbalances in papillary tumors. Since then we could identify 17 published papers on 48 patients. [3],[4],[5],[6],[7],[8],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18],[19],[20],[21] A review of the literature revealed that there was no uniform consensus in the management of these lesions, which may be related to the smaller number in each treatment category [Table - 1], and that none of the prognostic variables reached statistical significance. The only variable that tended to be associated with a better overall survival and recurrence was the extent of resection. [10] Most of the tumors recurred during the follow-up period necessitating radiation, chemotherapy, or surgical resection. It is not yet clear as to which is the best initial management strategy or how to manage a recurrent tumor. In some of the earlier published cases of PTPR, the initial pathologic diagnosis was different. Of the 15 cases with features of PTPR studied by Hasselblatt et al., [9] only 3 cases had been filed as PTPR and the remaining 12 cases with different pathologic diagnosis. In such cases, the initial management strategy and management of the residual lesion might have been dictated by the pathology report. In one of the published cases, the tumor was initially diagnosed as metastatic adenocarcinoma. [12] Our patient was initially treated at other facility with a ventriculoperitoneal shunt and empirical gamma knife radiosurgery. The reason for such an approach was not clear. Initially, however, this had a very good response, but he had a recurrence 7 years later. Ours is the first case of a PTPR managed with gamma knife radiosurgery initially with a good response. Earlier, stereotactic radiosurgery had been used as an adjuvant treatment in 3 cases [3],[12] and in the salvage treatment for one recurrence [10] [Table - 1]. With just one case managed with gamma knife radiosurgery, we cannot recommend it as an initial treatment option. Probably in the treatment of PTPR, gamma knife radiosurgery can be an option. Whether radiosurgery alone or some combination of treatment with chemotherapy and surgery is best remains to be defined, but the outcome of treatment seems to rely most heavily on the histologic diagnosis. The prognostic categories of pineal tumors may be divided into germinomatous or nongerminomatous, which include teratomas, choroid plexus tumors, ependymomas, parenchymal tumors, such as pineocytomas and pineoblastomas. Jouvet et al., [3] based on the histopathologic findings, concluded that these tumor cells may be derived from the ependymal cells of the subcommissural organ. The papillary tumor, since it is presumed to be of ependymal origin, is nongerminomatous. The distinction between germinomatous and nongerminomatous tumors makes a great difference in the primary treatment approach. Localized germinomas can be managed with radiation alone. [22] Yen et al.[23] had reported that additional chemotherapy had no significant effect on survival but was associated with a higher incidence of treatment-related toxicity. For germinomas, a radiation dose of 25 Gy to ventricular system or whole brain and 25 Gy tumor boost is usually administered. [22] With external beam radiation therapy alone, the 5-year survival rate for germinomatous tumors of the central nervous system has been reported anywhere between 75% and 100% with a recurrence rate of 10%-17% usually within 2 years after irradiation. [24] For germinomatous tumors with gamma knife radiosurgery, a local tumor control rate of 82% at 3 and 5 years was reported by Mori et al.[25] Kobayashi et al. reported a higher control rate (100%) with gamma knife radiosurgery for germinomatous tumors. [26] In the above-reported studies, [25],[26] in most of the cases, gamma knife radiosurgery was not administered as a primary modality but as an adjunctive to radiation, craniotomy, or chemotherapy, hence the results need to be interpreted cautiously. For pineal tumors, the marginal dose (50% isodose) varied between 9.9 and 25.7 Gy. [25] Nongerminomatous germ cell tumors are resistant to radiotherapy, but respond favorably with systemic management, such as chemotherapy. [27],[28] For PTPR, Dagnew et al.[12] reported a dose of 15 Gy (85% isodose line) in 2 of their cases where radiosurgery was used as an adjunctive treatment 3 weeks after the surgery. In the remaining cases where SRS (Stereotactic Radiosurgery) was used, there was no documentation of the radiation dose. Radiation treatment for PTPR was used primarily as an adjunctive treatment. It was also used after a local recurrence. Buffenoir et al.[16] in their patient had administered a localized radiotherapy to the tumor bed at a dose of 50 Gy in fractionated doses over a period of 5 weeks. In the case by Santarius et al., 55 Gy in 33 fractions was administered to the tumor region. [19] The radiation dose was based on the assumption that the tumor was a choroid plexus carcinoma. In the case by Dagnew et al., [12] the patient underwent adjunctive whole-brain radiotherapy (30 Gy in 10 fractions) 2 weeks after surgical resection. References
Copyright 2010 - Neurology India The following images related to this document are available:Photo images[ni10120f5.jpg] [ni10120f3.jpg] [ni10120f1.jpg] [ni10120f7.jpg] [ni10120f2.jpg] [ni10120f6.jpg] [ni10120f4.jpg] [ni10120t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}