 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
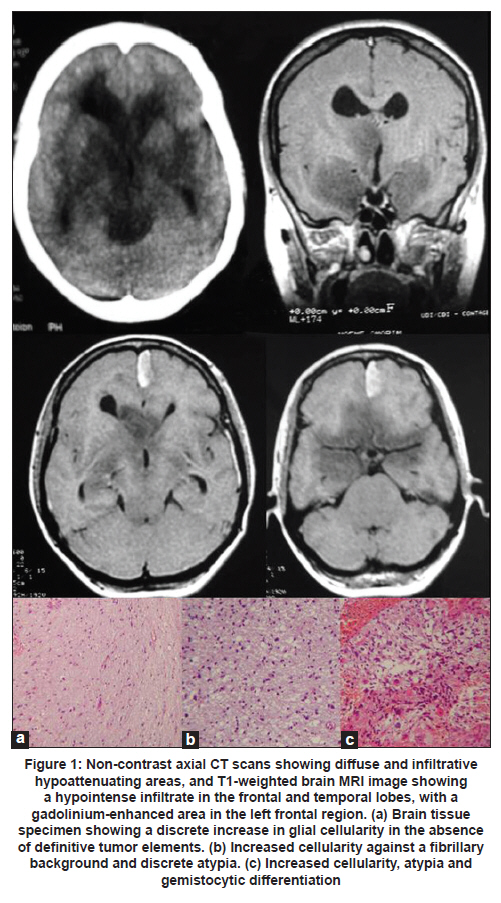
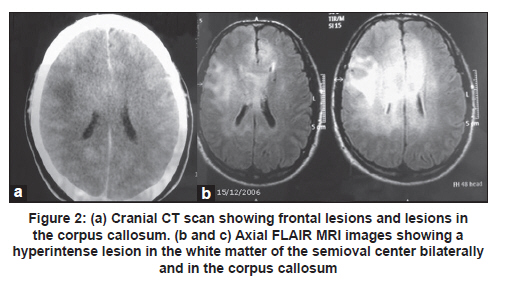
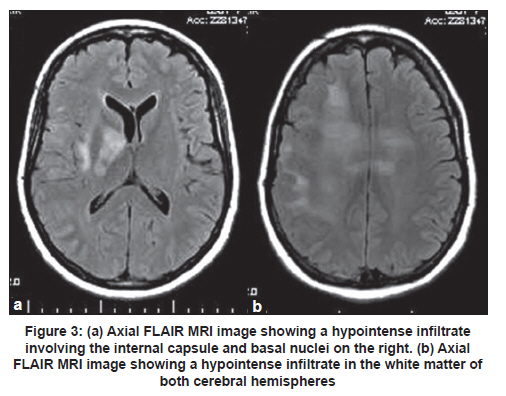
Neurology India, Vol. 59, No. 1, January-February, 2011, pp. 122-125 Case Report Gliomatosis cerebri: Diagnostic considerations in three cases Rafael Augusto Castro Santiago Brandão1, Gervásio Teles C. de Carvalho1, Camilo Augusto de Azeredo Coutinho2, Paulo Pereira Christo3, Cesar Felipe Gusmão Santiago4, Maria do Carmo Vasconcelos Santos3, Atos Alves de Sousa1 1 Department of Neurosurgery, Neurosurgeon of Santa Casa Hospital, Faculty of Medical Science of Minas Gerais, Belo Horizonte, Brazil Clinical symptoms and radiologic characteristics of gliomatosis cerebri (GC) are non-specific and the condition may be confused with other central nervous system diseases. We report three patients with GC; all the three patients had involvement of more than three lobes and the deep white matter, as well as bilateral involvement. Differentiation of GC from other neurologic diseases involving diffuse white matter may be difficult. However, the diagnosis can be based on the combination of radiologic and histopathologic features. Keywords: Demyelinating brain disease, glial tumors, glioma, gliomatosis cerebri Introduction World Health Organization (WHO) defines gliomatosis cerebri (GC) as a distinct nosological entity among other glial tumors of the central nervous system (CNS). Gliomatosis cerebri is classified as a diffusely infiltrating neuroepithelial tumor, which involves at least two cerebral lobes and, occasionally, infratentorial structures or the spinal cord. The brain architecture is commonly preserved, with neurons being spared, and the mass effect is minimal. [1],[2] Clinical symptoms and radiologic characteristics are non-specific and the condition may be confused with other CNS diseases. The presenting clinical features include: headache, focal deficits, cognitive and behavioral abnormalities, features of elevated intracranial pressure, and epileptic seizures. [3] We report three patients with GC and present a short review of the literature. Case Reports Case 1 A 39-year-old woman presented with a four month history of mental confusion, partial complex seizures, and urinary incontinence. Cranial computed tomography (CT) scan demonstrated multifocal hypodense periventricular and posterior fossa lesions. Magnetic resonance imaging (MRI) of the brain revealed diffuse infiltrative lesions in the temporal and frontal lobes and thalami, hypointense on T1-weighted sequences and hyperintense on T2-weighted and FLAIR sequences. Contrast study showed a small area of contrast uptake in the left frontal region [Figure - 1]. The diagnostic hypothesis was GC. Investigation of oncotic cells in cerebrospinal fluid (CSF) and serology for HIV were negative. Histopathology of stereotactic biopsy of the left frontal diffuse lesion showed a discrete increase in cellularity and mild autolysis. There were no features to suggest high-grade glioma or metastatic tumor. Since this biopsy was inconclusive, repeat stereotactic biopsy was performed, one from the left perithalamic region, two from the left periventricular region, and one from the frontomedial contrast uptake region on the left. Histopathologic analysis showed increased cellularity, atypia, and gemistocytic differentiation in one of the frontal fragment, findings consistent with grade III glioma. The periventricular fragment showed an increase in cellularity against a fibrillary background associated with discrete atypia, compatible with a low-grade glioma (grade II fibrillary astrocytoma). The thalamic fragment showed an increase in glial cellularity in the absence of definitive tumor elements [Figure - 1]a-c. Taken together, the imaging findings and the results of the anatomopathologic analysis led to a diagnosis of GC. The patient was submitted to radiotherapy but died about 10 months after the onset of symptoms. Case 2 A 51-year-old man presented with mild daily holocranial headache with no other symptoms of 40 days duration. The neurologic examination was normal. Cranial CT scan showed isodense lesion involving the cerebral cortex and the adjacent white matter of the centrum semioval. The lesion was centered on the trunk of the corpus callosum, resulting in attenuation of the frontal horns, and another hypodense lesion involving the subcortical white matter of the right frontal region, without contrast enhancement [Figure - 2]a. Brain MRI showed an extensive diffuse infiltrative lesion with no mass effect, hypointense on T1-weighted sequences and hyperintense on FLAIR sequences, involving the corpus callosum and white matter of the semioval center bilaterally [Figure - 2]b and c. Cerebrospinal fluid analysis revealed: protein 51 mg/dL, glucose 65 mg/dL, leukocytes 0/mm 3 , red blood cells 1/mm 3 , negative Gram staining, and a non-reactive VDRL test. Direct investigation for fungi and alcohol acid-resistant bacilli (AARB) was negative. Culture for bacteria, fungi, and AARB was negative. Protein electrophoresis showed no alterations, and no oligoclonal bands were observed. Tests for herpes simplex virus 1 and 2 were negative. Blood tests: anti-HIV 1 and 2 negative, non-reactive VDRL and antinuclear factor (ANF) negative. Histopathology of the stereotactic biopsy from the corpus callosum on the right showed features suggestive of gliosis. Immunohistochemistry showed a gemistocytic pattern of astrocytic proliferation consistent with the diagnosis of grade II astrocytoma and, together with the imaging findings, the diagnosis of GC was made. The patient was started on oral corticosteroids and was submitted to radiotherapy. At 25-month follow-up the patient had dizziness but no headaches. Neurologic examination showed discrete gait ataxia. Case 3 A 50-year-old woman, a known case of seizure disorder associated with neurocysticercosis in remission and off antiepileptic drugs, presented with a 20-day history of headache and left central facial paralysis progressing to left hemiparesis and generalized tonic-clonic seizures. Cranial CT scan showed discrete attenuation of the gyri and sulci of the right cerebral hemisphere. FLAIR and T2-weighted brain MRI revealed a hyperintense area in the left and, mainly, in the right cerebral hemisphere [Figure - 3]a and b. CSF analysis showed: leukocytes 8/mm 3 (96% lymphocytes), protein 38 mg/dL, and glucose 99 mg/dL. Gram staining, AARB and China ink preparation, and VDRL were negative. Initial diagnosis considered was herpes encephalitis. She was started on acyclovir and discontinued six days later after CSF PCR was negative for HSV 1/2. Histopathology of the stereotactic brain biopsy revealed cells with pleomorphic and hyperchromatic nuclei with some mitotic figures and focal hemorrhage in the fibrillary stroma. No necrosis or endothelial proliferation was observed. Some fragments were completely occupied by the lesion, whereas others showed infiltration of nervous tissue by atypical cells. These findings were suggestive of grade III astrocytoma. These results, together with the imaging findings, were compatible with GC. Chemotherapy with temozolomide was indicated and the patient was submitted to one radiotherapy session. However, the patient presented worsening of muscle strength on the left side and seizures. A cranial CT scan showed an increase in the size of the tumor with intratumoral bleeding. The patient developed right hemiplegia, aphasia, and progressive worsening of sensorium, and died. Discussion Gliomatosis cerebri is a rare CNS lesion and before the advent of MRI it was diagnosed only at autopsy [4] The term GC was first proposed by Nevin in 1938. [5] Since then, just over 300 cases have been reported in the literature. [2],[6],[7],[8],[9],[10],[11],[12],[13],[14],[15],[16],[17],[18],[19] The largest case series was by Taillibert et al. [3] who analyzed 90 cases from the French neuro-oncology network between 1993 and 2004 and more than 206 cases described in the literature between 1938 and 2004. Gliomatosis cerebri is an infiltrative diffuse glial tumor but not destructive lesion, and brain architecture is frequently preserved. [1] This lesion was included as a distinct nosological entity among neuroepithelial tumors in 2007. These tumors can be classified into two types: primary GC (type 1) in which no pre-existing primary cerebral neoplastic lesion is diagnosed, and secondary GC (type 2) in which a pre-existing cerebral neoplasm progresses and infiltrates other cerebral lobes. Most common type is type 1, [2] similar to the three patients described here. Involvement of more than three lobes and of the deep white matter, as well as bilateral involvement, was the common findings among all the three patients. The sites most frequently involved by GC are: basal and thalamic nuclei (75%), corpus callosum (50%), brainstem and spinal cord (10-15%), and cerebellum (10%), with two or more sites generally being affected at the same time. [4] In a review of 296 cases, the most frequent tumors were the astrocytic (n = 108), oligodendroglial (n = 54), mixed (n = 17), and indeterminate type (n = 117) [3] Owing to the infiltrative nature of GC, blood supply of the tumor is by pre-existing cerebral vessel, not requiring the formation of new vessels. Almost no angiogenesis is observed in GC, and this fact is demonstrated by analysis of the tumor microvasculature, which maintains the immunohistochemical characteristics of the blood-brain barrier. As a consequence, the tumor generally does not take up contrast. [20] The incidence of GC is higher among men than women (1.31), with a mean age of diagnosis at 39 and 45 years, respectively. [3] The youngest patient reported was one-month-old girl and the oldest patient was 85-year-old man. [3] The clinical presentation is determined by the location of the tumor, and the clinical features in decreasing order of frequency include: seizures, intracranial hypertension, behavioral and cognitive abnormalities, and focal neurologic deficits. [4] Headache may occur, as observed in two of the patients, but no characteristic pattern of pain has been described. [21] Recently, two patients with GC who manifested with Parkinsonian symptoms were described. [9],[12] The cranial CT scan findings are non-specific and include loss of differentiation between gray and white matter and asymmetric hypodensities, and CT can be normal in some cases. [4] Brain MRI may show a homogenous infiltrative lesion which is isointense or hypointense on T1-weighted sequences and hyperintense on T2-weighted and FLAIR sequences, with minimum uptake of gadolinium on T1-weighted images. [5] However, these are non-specific characteristics. The diagnosis of GC is based on the combination of radiologic and histopathologic evidence. Hyperintensity on T2-weighted images reflects tumor infiltration, but may also represent secondary destruction of myelin fibers which is typically observed in the white matter, corpus callosum, basal ganglia and thalamus, but is rare in the brainstem, cerebellum, and spinal cord. [22] The observation of bilateral white matter alterations in the presence of a diffuse and infiltrative tumor involving the basal ganglia, thalamus, corpus callosum and, less frequently, the brainstem and hypothalamus is highly suggestive of GC. The presence of contrast uptake may suggest worse prognosis. [2] The differentiation between GC and other neurologic diseases that diffusely involve the white matter, such as progressive multifocal leukoencephalopathy, multiple sclerosis, acute disseminated encephalomyelitis, viral encephalitis, CNS vasculitis, Behçet's disease, cerebrovascular diseases and venous sinus thrombosis, is difficult, but MR spectroscopy can be of help. [18],[22] In patient 3, a brain biopsy led to the suspicion of herpes encephalitis and empirical treatment with acyclovir was initiated. In Cases 1 and 2, the differential diagnosis was diffuse demyelinating disease. The prognosis of GC is poor, with a median survival of 14.5 months. Male patients, younger patients, and those with a tumor of the oligodendroglial type have a better prognosis. [3] In the case of children, there are isolated reports of stable progression from 6 to 19 years after chemotherapy and radiotherapy. However, a study involving 13 patients aged 1.6 to 18 years showed an overall survival rate of 64% after 2 years. [2] Current treatment of GC is based on total brain irradiation and chemotherapy. Chemotherapy includes the use of procarbazine and vincristine or oral temozolomide, with the last drug being less toxic. Radiotherapy and chemotherapy can reduce or stabilize the tumor and improve the symptoms of GC, but they do not change patient survival. [23] However, the outcomes are variable because of variations in the pathologic spectrum of the disease. References
Copyright 2011 - Neurology India The following images related to this document are available:Photo images[ni11032f1.jpg] [ni11032f3.jpg] [ni11032f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}