 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
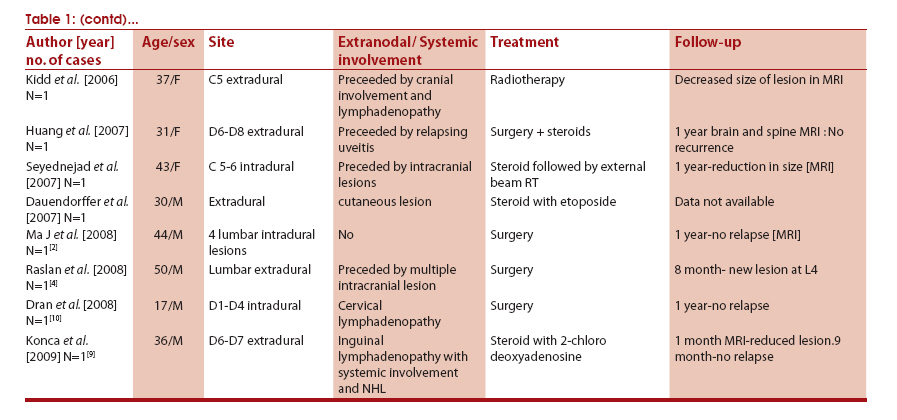
Neurology India, Vol. 59, No. 3, May-June, 2011, pp. 438-442 Case Report Rosai-Dorfman disease presenting as cervical extradural lesion: A case report with review of literature Tanmoy K Maiti1, Jagathlal Gangadharan1, Anita Mahadevan2, A Arivazhagan1, BA Chandramouli1, SK Shankar2 1 Department of Neurosurgery, National Institute of Mental Health and Neurosciences, Bangalore, India Correspondence Address: Jagathlal Gangadharan Department of Neurosurgery, National Institute of Mental Health and Neurosciences, Bangalore-560 029, Karnataka India drjagathlal@yahoo.com Date of Submission: 28-Sep-2010 Code Number: ni11126 PMID: 21743179 DOI: 10.4103/0028-3886.82769 Abstract Sinus histiocytosis with massive lymphadenopathy or Rosai-Dorfman disease is a rare, but well documented entity. We report a lady who presented with progressive quadriparesis, with cervical extradural lesion on magnetic resonance imaging. She underwent decompression of the lesion and histological diagnosis of the lesion was Rosai-Dorfman disease. On one-year follow-up, she had complete improvement of the deficits with no further progression of the lesion. The presentation of this disease as an isolated spinal extradural mass lesion is quite rare, with only six cases reported in literature.Keywords: Rosai-Dorfman disease, sinus histiocytosis with massive lymphadenopathy, spinal extradural lesion, steroids, surgery Introduction Rosai-Dorfman disease (RDD) is a rare, benign, self-limiting lymphoproliferative disorder first described by Rosai and Dorfman in 1969. [1] Typical clinical presentation is with massive painless cervical lymphadenopathy, but extranodal involvement is rare, seen in 30 - 40% of the cases, with skin, upper respiratory tract, orbit and eye, salivary gland, bone, and testis being reported. [1] In less than 5% of the cases, the central nervous system (CNS) is involved. [2] Involvement of the spine has been described in only 20 - 25% of those with CNS involvement [3],[4] and is generally not accompanied by lymphadenopathy. [3] We report a case of RDD presenting with extradural spinal cord compression, and briefly review the literature. Case Report A 19-year-old lady presented with a two-month history of an insidious onset of gradually progressive neck pain and asymmetric progressive spastic quadriparesis without any sphincter dysfunction. Neurological examination revealed hypertonia of both lower limbs. She had motor power of 2/5 in the right side and 3/5 in the left side, with decreased sensation, below C5. Deep tendon reflexes were brisk and bilateral plantar response was extensor. There was no lymphadenopathy. A complete hemogram revealed hemoglobin of 10.8 gm/dL, total WBC count of 13,600/cu mm, and ESR of 62 mm /1 hour. Serum levels of immunoglobulin A, G, and M were normal. An ultrasound of the neck and abdomen did not reveal any abnormality. Magnetic resonance imaging (MRI) of the cervical spine revealed a posteriorly placed, extradural lesion at C3 - C6, with contrast enhancement and doubtful intradural encroachment [Figure - 1]. She underwent C2 - C6 laminectomy and excision of the extradural lesion. The lesion was moderately vascular, fleshy, and adherent to the dura, extending from C2 - C7. The dura was freed from the lesion, and was lax and pulsatile after decompression. Histopathology revealed infiltration of the extradural soft tissue by dense aggregates of lymphocytes and histiocytes [Figure - 2]a. Emperipolesis with intact lymphocytes within the cytoplasm of the large cells were present in the infiltrate [[Figure - 2]a, inset]. Also seen were clusters of large histiocytes with eosinophilic, finely granular cytoplasm, and multinucleate giant cells [Figure - 2]b that demonstrated strong immunoreactivity for S100 [[Figure - 2]b, inset top right] and CD68 [[Figure - 2]b, inset bottom left]. The cells were immunonegative for CD1a excluding the Langerhans cell histiocytic lesion. Postoperatively the patient improved gradually with physiotherapy and became ambulatory in three weeks and returned to work within six weeks. At the 18-month, follow-up, she did not have any neurological deficits. There was no evidence of other extranodal or lymph node involvement. A one-year postoperative, follow-up MRI, showed doubtful residue/recurrence opposite C4. The patient did not receive any steroid, radiotherapy or chemotherapy. Discussion CNS Rosai-Dorfman disease can present at any age, but most commonly in the third and fourth decade, with a slight male predominance. [1],[5] Over 90% of the CNS Rosai-Dorfman involve the leptomeninges and seen on neuroimaging as a dural-based, contrast-enhancing lesion, mimicking a meningioma. [2],[3],[5] Although thought to be benign, reversal of neurological deficits is mostly incomplete and deaths have been reported due to infiltration of the vital organs. [1] The etiology of RDD remains largely unknown. It is presumed to be reactive in nature, as no evidence of clonality has been demonstrated by molecular analysis. A possible association with infective agents like HHV-6 and Parvo B19 have been suggested in a few studies. [6] The current hypothesis is that RDD is a subtle, yet undefined immunological defect, which promotes monocyte recruitment from the circulation into the nodal or extranodal sites followed by transformation into the immunophenotypically distinct RDD histiocytes demonstrating emperipolesis with functional uniqueness in terms of the cytokine expression profile. [7] Cytologically, emperipolesis is a consistent finding in nodal disease, although less apparent in extranodal sites, whereas, fibrosis is commonly seen. [5] Emperipolesis refers to phagocytosis of intact lymphocytes by macrophages (also known as lymphocytophagocytosis). [5] It differs from phagocytosis, in that the lymphocytes taken up are not attacked by the enzymes and appear intact within the histiocytes. A biopsy remains the gold standard for diagnosis and the differential diagnoses include hematopoietic and primary CNS lesions that are accompanied by dense inflammatory infiltrates, such as, lymphoplasmacytic meningioma, Langerhans histiocytosis, nodular sclerosing variant of Hodgkin's disease, intracranial plasmacytoma, and plasma cell granulomas. Immunohistochemistry aids in differentiating these from the other Langerhans and non-Langerhans cell histiocytosis. The possibility that many of the earlier reported cases of CNS plasma cell granulomas or inflammatory pseudotumors are RDD, has also been suggested. [8] There is no established treatment protocol for systemic RDD as it is often self-limiting except in the event of the compression of vital organs or airway obstruction. Local recurrence is possible following surgical resection. [1],[4] Steroids often resolve fever and reduce lymph node size. The role of radiotherapy and chemotherapy is controversial. [9] A thorough literature search revealed at least 35 previously reported cases with spinal involvement, as depicted in the [Table - 1a & b], although isolated spinal involvement was seen in only six of them. Analyzing them, there is a male predominance [21:11] with the age group varying from two-and-a-half years to 78 years with a mean age of 31.8 years. Simultaneous nodal involvement was found in only ten cases. Cranial involvement was found in six cases and the highest interval between cranial and spinal involvement was five years. Extra-CNS involvement was found in six patients. Lesion were most prevalent at the thoracic level (12 cases) followed by the cervical level (six cases); three were found in the cervicodorsal region; two each in the lumbar and sacral levels; and one in the craniovertebral junction. The lesions were multiple in five cases. The plane of the lesion was primarily extradural in 18 cases, whereas, eight were intradural and three were intramedullary. In three cases, the lesions were primarily involving the body with extradural extension. Surgery was the treatment of choice in most cases, for relieving cord compression. Recurrence was noted in three cases after surgery, all within one year. [4] In eight cases where chemotherapy, radiotherapy or their combination was preferred over surgery, the control of disease was possible, although the reversal was not complete. Two patients expired because of multiple organ involvement. [1] In our case, a significant challenge was encountered in diagnosing as well as formulating a further line of treatment, in view of the exclusive extranodal involvement of this disease, which resulted in rapidly progressive quadriparesis. Early diagnosis of the extranodal intraspinal RDD could help to avoid a permanent long-term neurologic sequel only if there is a high index of suspicion on the part of clinicians and pathologists, of this very rare cause of spinal cord compression.[10] References
Copyright 2011 - Neurology India The following images related to this document are available:Photo images[ni11126f2.jpg] [ni11126t1b.jpg] [ni11126t1a.jpg] [ni11126f1.jpg] |
| |||||||||

![[Figure - 1]](/showimage?ni/photo/ni11126f1.jpg){kind=link}
![[Figure - 2]](/showimage?ni/photo/ni11126f2.jpg){kind=link}
{kind=link}
{kind=link}