 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
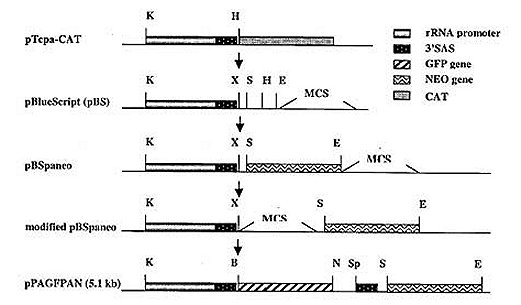
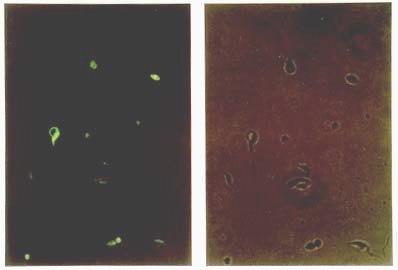
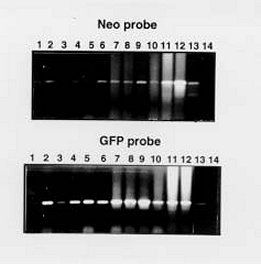
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol. 95(1): 111-114, Jan./Feb. 2000 SHORT COMMUNICATION Colony Polymerase Chain Reaction of Stably Transfected Trypanosoma cruzi Grown on Solid Medium Wagner G dos Santos++, Ivelina Metcheva, Gregory A Buck+ Department of Microbiology and Immunology, Medical College of Virginia Campus, Virginia Commonwealth University, Box 980678 Richmond, VA 23298-0678, USA + Corresponding author. Fax: (804) 828-9946. E-mail: buck@hsc.vcu.edu.++ Current address: Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Rua da Quinta Grande nº 6, Apartado 127, Oeiras, 2780-Portugal.Received 18 August 1999 Code Number:OC00018 Tools for the genetic manipulation of Trypanosoma cruzi are largely unavailable, although several vectors for transfection of epimastigotes and expression of foreign or recombinant genes have been developed. We have previously constructed several plasmid vectors in which recombinant genes are expressed in T. cruzi using the rRNA promoter. In this report, we demonstrate that one of these vectors can simultaneously mediate expression of neomycin phosphotransferase and green fluorescent protein when used to stably transfect cultured epimastigotes. These stably transfected epimastigotes can be selected and cloned as unique colonies on solid medium. We describe a simple colony PCR approach to the screening of these T. cruzi colonies for relevant genes. Thus, the methodologies outlined herein provide important new tools for the genetic dissection of this important parasite. Key words: Trypanosoma cruzi - stable transfection - Green Fluorescent Protein - neomycin phosphotransferase Molecular studies of kinetoplastid parasites have revealed many unique mechanisms of gene regulation, gene expression and RNA processing in these organisms (Borst 1986, Vanhamme and Pays 1995). However, a general lack of efficient DNA transfection systems has precluded genetic dissection of these novel genetic processes. The development of transfection systems as a tool for genetic analysis of protozoan parasites has initiated a new phase in the study of these organisms, and both transient and stable transfection systems have been successfully employed to introduce and express foreign or modified genes. Among other applications, these transfection systems have been used: (i) to identify and characterize transcription promoters such as the rRNA and SL gene promoters in Trypanosoma cruzi (Tyler-Cross et al. 1995, Nunes et al. 1997a,b) and the PARP and VSG promoters in T. brucei (Zomerdijk et al. 1990, Rudenko et al. 1990), (ii) to functionally complement selected endogenous genes (Kelly et al. 1994, Nozaki & Cross 1994) and, (iii) to analyze gene function based on inactivation by targeted gene disruption (Lee & Van der Ploeg 1990, Otsu et al. 1993, Cooper et al. 1993, Chung et al. 1994). In many cases after transfection of the parasite, it is necessary to select and clone the transformants before further analysis. For T. cruzi, this cloning has been done primarily by the extremely time consuming and uncertain process of limiting dilution and subculture in liquid medium. Only recently, an optimized method to isolate transformants from solid medium was reported (Mondragon et al. 1999). Herein, we report a simple method to select stable transfected T. cruzi epimastigotes grown on solid medium and to analyze the genotype by using colony polymerase chain reaction (PCR). We have constructed a plasmid, pPAGFPAN, which contains the T. cruzi rRNA promoter cloned upstream from the green fluorescent protein (GFP) gene from the jellyfish Aequorea victoria (Kain et al. 1995) and the bacterial neomycin phosphotrans-ferase gene (NEO) (Fig. 1). To be properly processed by trans-splicing the splice acceptor sites 35.1 SAS and 35.2 SAS (McCarthy-Burke et al. 1989) were inserted upstream of NEO and GFP genes, respectively. Transfection of T. cruzi epimastigotes strain CL-brener with this plasmid generated a neomycin resistant (neor) population, which contained parasites co-expressing GFP (Fig. 2). After plating these parasites in blood-agar-LIT solid medium supplemented with 300 µg/ml of G418 and incubating the plate at 28°C for 20-30 days, single isolated ~1 mm2 opaque colonies appeared (Fig. 3). Although the plating efficiency of these transfected parasites was quite low (~0.005), these colonies represent clones of the stably transfected T. cruzi. Fig. 1: construction of the plasmid pPAGFPAN. Initially the 654 nt insert which contains the Trypanosoma cruzi rRNA promoter and the 35.1 SAS was excised from pTcpaCAT (Tyler-Cross et al. 1995), gel purified and ligated into KpnI (K), HindIII (H) linearized pBlueScript phagemid (Stratagene, La Jolla, CA). The 800 nt NEO gene fragment was PCR amplified from pSV2neo plasmid using the primers set: forward 5’GTCGACATGATTGAACAA3’and reverse: 5’AGAAGAACTCGTCAAGAA3’ under the following cycling conditions: 94°C denaturation for 1 min, 50°C annealing for 30 sec and 72°C extension for 1 min. This fragment was cloned downstream the SAS into the SalI (S) and EcoRI (E) sites located in the MCS of the phagemid (pBSPANEO). The remaining restriction sites from this region, located downstream from the NEO fragment, were excised and a new MCS containing the sites XbaI (X), BglII (B), StuI, AvrII, SacI, NdeI (N), SpeI (Sp) and SalI was synthesized as complementary oligonucleotides and cloned into XbaI and SalI sites between the 35.1 SAS and the NEO gene (modified pBSpaneo). Since the insertion of GFP upstream from the NEO gene would leave it without a splice acceptor, a new 35.2 SAS was synthesized as complementary oligonucleotides and cloned into the NdeI (N) and SpeI (Sp) sites. The 700 nt GFP gene was PCR amplified from pGFP (ClonTech Laboratories, Palo Alto, CA) using primer pair; forward 5’-GGGGACAGATTAATCGAATGCTATTTGTATAGTTCATC-3’and reverse 5’-GACTCTAGAGGATCCCCGGGT-3’under the cycling conditions: 94°C denaturation for 1 min, 60°C annealing for 30 sec and 72°C extension for 1 min, and cloned into BglII(B) and NdeI (N) compatible ends. The final plasmid pPAGFPAN was used in the transfections. All oligonucleotide synthesis and DNA sequencing required in these experiments was performed in the MCV-VCU nucleic acids research facility. Fig. 2: expression of GFP and neomycin phosphotransferase (NEO) in transfected Trypanosma cruzi. T. cruzi epimastigotes transfected with pPAGFPAN were selected by growth for 24 days in LIT medium containing 500 µg/ml of G418. Paraformaldehyde fixation was performed by washing cells twice in PBS and resuspending them at a density of 106 cells/ml in 4% paraformaldehyde/PBS for 1 h. These fixed cells were washed twice in PBS supplemented with 5% FCS and 10 µl were added onto a slide and visualized on a Olympus fluorescent microscope adapted with a standard filter set for fluorescein isothiocyanate (FITC). Photographs were taken with a 40X objective and Kodak ektachrome 1600 ASA film. Right panel, without UV light exposure, left panel, with standard fluorescein isothiocyanate (FITC) UV filter set. Fig. 3: colony PCR of Trypanosoma cruzi. Single colonies of T. cruzi transfected with pPAGFPAN and grown on solid medium blood/agar-LIT were picked and the extracted DNA used for PCR reaction using NEO (upper panel) or GFP (lower panel) primer pairs, described on legend of Fig. 1. Lanes 1-6, PCR products from single colonies that had been inoculated in liquid medium supplemented with 500 µg/ml of G418; lanes 7-12, PCR products from single colonies taken directly from the plates; lane 13, positive control; lane14, negative control. PCR cycling conditions for both GFP and NEO primer pair were 94°C denaturation for 1 min, 52°C annealing for 30 sec and 72°C extension for 1 min. To verify the presence of the reporter gene (GFP) in these T. cruzi colonies, colony PCR was performed. Several colonies were picked using a sterile plastic pipette tip, and transferred to 20 µl of sterile double distilled H2O. These T. cruzi suspensions were incubated at 100°C for 5 min, vortexed vigorously for 2 min to release the nucleic acids, and the cellular debris was removed by centrifugation for 2 min at top speed in a microcen-trifuge. The supernatant was removed and transferred to a clean microcentrifuge tube and 10 µl was used as DNA template in PCR reactions with GFP and NEO-specific primers (Fig. 3) flanking either the NEO or GFP gene. All colonies examined showed a 700 bp PCR fragment derived from the GFP gene and a 800 bp fragment derived from the NEO gene (Fig. 3). At the same time the colonies were picked for PCR analysis, an aliquot was also picked to inoculate 5 ml of LIT medium (Camargo 1964) supplemented with inhibitory concentrations (500 µg/ml) of G418. These cultures were incubated for seven days, and 1 ml was pelleted by centrifugation in a microcentrifuge tube. The pellet of T. cruzi epimastigotes was resuspended in 20 µl of sterile water and subjected to PCR analysis as described above. PCR product with size consistent with that expected for NEO and GFP genes were observed, confirming the presence of the plasmid in the cultured parasites (Fig. 3). This work provides an important tool for the genetic manipulation of T. cruzi and for monitoring the genotype of transfected T. cruzi. Thus, transfected epimastigotes were selected and cultured as clonal colonies on solid medium supplemented by inhibitory concentrations of G418. This approach for cloning transformed T. cruzi represents significant advantages of time and effort over the limited dilution approach previously available, and will facilitate more the ongoing genetic dissection of these important pathogens. This work was supported by grants from the National Institutes of Health, the American Heart Association, and The Jeffress Memorial Trust. WGS was supported by a fellowship from CNPq. REFERENCES
Copyright 2000 Fundacao Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc00018b.jpg] [oc00018c.jpg] [oc00018a.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}