
|
Memórias do Instituto Oswaldo Cruz
Fundação Oswaldo Cruz, Fiocruz
ISSN: 1678-8060 EISSN: 1678-8060
Vol. 97, Num. 1, 2002, pp. 109-111
|
Mem Inst Oswaldo Cruz, Rio de
Janeiro, Vol. 97(1) 2002, pp. 109-111
SHORT COMMUNICATION
A Heminested Polymerase Chain
Reaction for the Detection of Brazilian Rabies Isolates from Vampire Bats and
Herbivores
RM Soares++, F Bernardi*, SM Sakamoto**/++,
MB Heinemann**, A Cortez**, LM Alves**/++, AD Meyer+++,
FH Ito**, LJ Richtzenhain**/+
Instituto de Ciências Biomédicas,
Universidade de São Paulo, São Paulo, SP, Brasil *Faculdade de
Veterinária, Universidade Santo Amaro, São Paulo, SP, Brasil **Faculdade
de Medicina Veterinária e Zootecnia, Universidade de São Paulo,
Av. Prof. Dr. Orlando Marques de Paiva 87, Cidade Universitária, 05508-900
São Paulo, SP, Brasil
+Corresponding author. Fax: +55-11-3818-7928. E-mail: leonardo@usp.br
++Fapesp fellowship
+++ Capes fellowship
Received 6 March 2001
Accepted 26 September 2001
Code Number: oc02020
A heminested-PCR (hn-PCR) using primers to
the nucleoprotein-coding gene in a nested set was evaluated in the detection
of Brazilian strains of rabies virus (RV). A representative number of RV nucleoprotein
sequences belonging to genotype 1 were aligned. Based on such alignment, primers
were directed to highly conserved regions. All 42 clinical samples positive
by both fluorescent antibody and mouse inoculation tests were also positive
by the hn-PCR. Brain tissue that had been left to decompose, obtained from an
experimentally inoculated mouse was tested by hn-PCR and yielded positive results.
In conclusion, primers designed here were capable of amplifying Brazilian RV
isolates obtained from a rural epidemiological cycle.
Key words: rabies virus - polymerase chain reaction
- detection of viral RNA - rabies diagnosis - Brazil
Rabies is a widespread zoonosis, which has been
of great concern due to its ability to determine a fatal acute encephalomyelitis
when the host is bitten by an ill animal (Wilkinson 1988). The causing agent
is a virus that belongs to the Lyssavirus genus in the Rhabdoviridae
family. Based on phylogenetic analyses of the nucleoprotein-coding gene (N gene),
the Lyssavirus genus has been divided into seven genotypes. Genotype
1 includes rabies virus (RV) strains (Bourhy et al. 1993). RV genome is composed
of a single-stranded, negative-sense, non-segmented RNA that codes for five
separate proteins: nucleoprotein, glycoprotein, phosphoprotein, membrane protein
and polymerase (Tordo 1991).
The World Health Organization (WHO) recommends
that the fluorescent-antibody test (FAT) and mouse inoculation test (MIT) carried
out simultaneously should be used for the detection of RV (Meslin et al. 1996).
FAT is a rapid and low cost technique, which may show positive results, as it
is able to detect viral antigens whether they are viable or not (Meslin et al.
1996). However, FAT's efficacy may be jeopardized when decomposed tissue is
used. In such cases, PCR may be used as a surrogate due to its more appropriate
performance (Sacramento et al. 1991, Kalmovarin et al. 1993, Whitby et al. 1997).
PCR based on N gene has been widely used for
diagnostic purposes since it is one of the most conserved fractions in RV (Smith
et al.1992, Kalmovarin et al. 1993, Heaton et al. 1997, Crepin et al. 1998,
Heaton et al. 1999, Black et al. 2000).
The aim of this study was to design primers based
on RV genotype 1 N gene and to evaluate the performance of a hn-PCR for direct
RV detection in clinical samples. Primers were based on the sequences of genotype
1 strains, including one Brazilian isolate (Kissi et al. 1995). Virus sequences
were aligned by using the Clustal X computer program (Thompson et al. 1997)
and the oligonucleotides were designed to recognize regions with high degree
of similarity within the N genes. The segments that were selected for primer
design are located in the middle of the N gene. Kissi et al. (1995) have demonstrated
that such gene fraction shows a striking level of similarity among RV isolates.
The physical and chemical properties of the primer were predicted by using the
Oligo 4.0 computer program (Rychlik & Rhoads 1989).
Primers with a maximum of three mismatches with
any aforementioned sequence and no mismatch in the 3' terminal nucleotide were
chosen. As the efficiency of PCR is inversely related to the length of the sequence,
primers were chosen based on the shortest flanking distance. Primer sets P510/P784
and P510/P942 (P510:ATA GAG CAG ATT TTC GAG ACA GC; P784:CCT CAA AGT TCT TGT
GGA AGA; P942:CCC ATA TAA CAT CCA ACA AAG TG) define 295 and 455 base pairs,
respectively.
Samples were 20% (w/v) homogenate of brain material
in PBS which had been stored at -20ºC and previously tested by FAT and
MIT (FAT/MIT), as prescribed by the WHO (Meslin et al. 1996). These samples
came from distinct geographical areas within the Brazilian territory and from
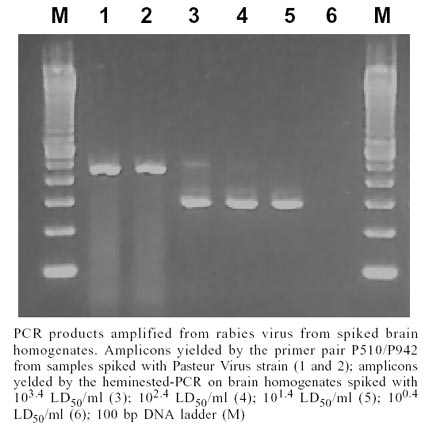
multiple species of herbivores and vampire bats (Table).
Total RNA was directly extracted from the samples by the TRIzol LS method, according
to manufacturer's instructions (Gibco BRL). Reverse transcription was performed
with 7 µl of the extracted product added to a final volume of 20 µl
containing 1 mM of each dNTP, 20 pmols of primer P510, 1 x RT buffer (Gibco
BRL), 1 mM dTT, 200 units of M-MLV reverse transcriptase (Gibco, BRL) and 0.01%
DEPEC treated ultra pure water. The mixture was incubated for 1 h at 42ºC.
A primary amplification was performed in 5 µl
of the reverse transcripted-cDNA template in a final volume of 50 µl,
containing 0.2 mM of each dNTP, 25 pmols of primer P510, 25 pmols of primer
P942, 1.5 mM of MgCl2, 1 x PCR buffer (Gibco, BRL), 1.25 units of
Taq DNA polymerase (Gibco, BRL) and ultra pure water. The amplification was
performed on a MJ Research PTC-200 Thermal Cycler. The heminested amplification
was performed in 5 µl of primary amplification template and primers P510
and P784. The following cycling conditions for the primary amplification were
adopted: initial heating at 94°C/3min, 35 cycles at 94°C/45 sec, 55°C/60
sec, 72°C/90 sec and a final extending step at 72°/10 min. The thermal
cycles for the nested assay were the same, except for an amplification phase
of 25 cycles. PCR products were run in 2% agarose gel electrophoresis in standard
TBE and stained with ethidium bromide 0.5 µg/ml (Sambrook et al. 1989).
Gels were observed under UV light and photographed.
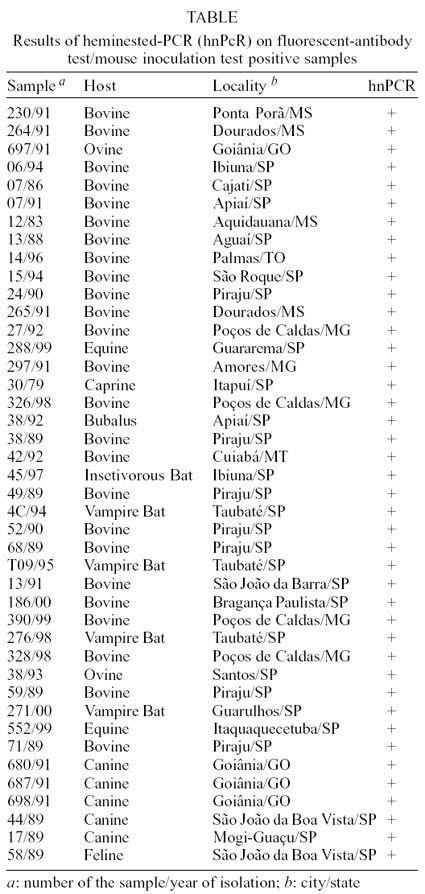
Amplicons yielded by both primer pairs are shown
in the Figure. The Table
shows hn-PCR results in samples from several Brazilian localities positive for
rabies by FAT/MIT. The ten samples which yielded negative results by FAT/MIT
also yielded negative results by PCR (results not shown). Total concordance
between hn-PCR and FAT/MIT was observed. Considering that other kind of rabies
virus may be found in rural environment, 5 canine and 1 feline rabid samples
were tested by the hn-PCR, yielding positive results in all cases.
Detection threshold for the hn-PCR method was
evaluated using normal brain homogenates of a mouse spiked with serial ten-fold
dilutions of Pasteur virus strain. Briefly, brain homogenates from experimentally
RV infected mouse containing 105.4 LD 50/ml were ten-fold serially
diluted with brain homogenates from a non-infected mouse. Titration of the brain
homogenates from the experimentally infected mouse was performed following the
method described elsewhere (Reed & Muench 1938). Brain homogenates containing
from 10 4.4 LD50/ml to 100.4 LD50/ml were tested by PCR.
The performance of hn-PCR on RV-infected brain
tissues at various stages of decomposition was also evaluated. Brain homogenates
from a mouse experimentally inoculated with PV strain were left at room temperature
for 24, 48, 72 and 96 h, and then tested by hn-PCR. The hn-PCR was able to detect
104.4, 103.4, 102.4 and 101.4 LD
50/ml (25000, 2500, 250 and 25 LD 50/ml). In the Figure,
the results of hn-PCR on 103.4, 102.4, 101.4
and 100.4 LD50/ml are shown. The hn-PCR was also capable of amplifying
RV genetic sequences from all the decomposed brain tissue samples which have
been left 24, 48, 72 and 96 h at room temperature.
False positive results are the main problem associated
with nested-PCR due to contamination with amplified DNA from the primary reaction
(Forghani & Erdman 1995). In order to avoid false positive results, some
extensive precautions should be taken: (i) each step of sample handling (RNA
extraction, first and second amplification steps) should be performed in different
laboratories; (ii) use of disposable gloves; (iii) analyses of no more than
ten samples per reaction; (iv) inclusion of negative control for each five samples.
The hn-PCR described in this paper was performed in three steps, one for cDNA
synthesis and two for both amplification assays.
As the detection of PCR products on agarose gel
may reveal spurious bands sometimes (results not shown), good candidates for
further improvements in the sensitivity, specificity and rapidity of the hn-PCR
are the combination of reverse transcription and first amplification in a single
step, and the standardization of methods other than agarose gel separation for
the detection of PCR products.
At present, simultaneous use of FAT and MIT is
irreplaceable for RV detection. However, their performance may occasionally
be impaired in decomposed tissues and/or samples containing no viable virus.
The goal of this study was to assess the fine
performance of the primers designed for Brazilian RV isolates obtained in rural
epidemiological cycle. Hn-PCR results presented here unequivocally provide an
additional tool for rabies diagnosis in some special cases, such as: (i) samples
which have been inadequately stored and then sent for laboratorial analysis;
(ii) when FAT alone yield negative results, FAT is impracticable to perform,
and results are urgent (for the entire hn-PCR procedure is accomplished in 24
h); and (iii) when dealing with small amount of sample as those obtained from
bats and laboratory animals in experimental studies.
REFERENCES
- Black EM, McElhinney LM, Lowings JP, Smith
J, Johnstone P, Heaton PR 2000. Molecular methods to distinguish between classical
rabies and the rabies-related European bat lyssaviruses. J Virol Methods
87: 123-131.
- Bourhy H, Tordo N, Kissi B 1993. Molecular
diversity of the Lyssavirus genus. Virology 194: 70-91.
- Crepin P, Audry L, Rotivel A, Gacoin C, Caroff
C, Bourhy H 1998. Intravitam diagnosis of human rabies by PCR using saliva
and cerebrospinal fluid. J Clin Microbiol 36: 1117-1121.
- Forghani B, Erdmann, DD 1995. Amplification
and detection of viral nucleic acids. In EH Lennette, DA Lennette, ET Lennette
(eds), Diagnostic Procedures for Viral, Rickettsial and Chlamydial Infections,
7th ed., American Public Health Association, Washington, p. 97-120.
- Heaton PR, Johnstone P, MCelhinney LM, Cowley
R, O'Sullivan E, Whitby JE 1997. Heminested PCR assay for detection of six
genotypes of rabies and rabies-related viruses. J Clin Microbiol 35:
2762-2766.
- Heaton PR, McElhinney LM, Lowings JP 1999.
Detection and identification of rabies and rabies-related viruses using rapid-cycle
PCR. J Virol Methods 81: 63-69.
- Kalmovarin N, Tirawatnpong T, Rattanasiwamoke
R, Panpanich T, Hemachuda T 1993. Diagnosis of rabies by polymerase chain
reaction. J Infec Dis 167: 207-210.
- Kissi B, Tordo N, Bourhy H 1995. Genetic polymorphism
in the rabies virus nucleoprotein gene. Virology 209: 526-537.
- Meslin FX, Kaplan MM, Koprowsky H 1996. Laboratory
Techniques in Rabies, 4th ed., WHO, Geneva, 476 pp.
- Reed LJ, Muench H 1938. A simple method of
estimating fifty per cent endpoints. Am J Hyg 27: 493-497.
- Rychlik W, Rhoads RE 1989. A computer program
for choosing optimal oligonucleotides for filter hybridization, sequencing
and in vitro amplification of DNA. N Acid Res 17: 8543-8551.
- Sacramento D, Bourhy H, Tordo N 1991. PCR
technique as an alternative method for diagnosis and molecular epidemiology
of rabies virus. Mol Cell Probes 5: 229-240.
- Sambrook J, Fritsch EF, Maniatis T 1989. Molecular
Cloning: a Laboratory Manual, 2nd ed., Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, 957 pp.
- Smith JS, Orciari LA, Yager P A, Seidel HD,
Warner CK 1992. Epidemiological and historical relationships among 87 rabies
virus isolates as determined by limited sequence analysis. J Infec Dis
166: 296-307.
- Thompson JD, Gibson T J, Plewniak F, Jeanmougin
F, Higgins DG 1997. The Clustal X windows interface: flexible strategies for
multiple sequence alignment aided by quality tools. N Acids Res 24:
4876-4882.
- Tordo N 1991. Contribution of molecular biology
to vaccine development and molecular epidemiology of rabies disease. Mem
Inst Butantan 53 (Supl.1): 31-51.
- Whitby JE, Johnstone P, Sillero-Zubiri C 1997.
Rabies virus in the decomposed brain of ethipian wolf detected by nested reverse
transcription-polymerase chain reaction. J Wild Disease 33:
912-915.
- Wilkinson L 1988. Understanding the nature
of rabies: an historical perspective. In JB Campbell, KM Charlton (eds), Rabie,
Kluwer Academic, Boston, p. 1-23.
© 2002
Instituto Oswaldo Cruz - Fiocruz
The following images related to this document are available:
Photo images
[oc02020f1.jpg]
[oc02020t1.jpg]
|

{kind=link}
{kind=link}