 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Mem Inst Oswaldo Cruz, Vol. 99, Suppl. 1, August, 2004, pp. 45-50 From Genomes to Vaccines Via the Proteome R Alan Wilson+, Rachel S Curwen, Simon Braschi, Stephanie L Hall, Patricia S Coulson, Peter D Ashton Department of Biology,
University of York, PO Box 373, York, YO10 5YW, UK Financial support: The Wellcome Trust, The Biology and Biotechnology Research Council, The European Commission INCO-DEV Programme, the WHO/UNDP/World Bank Programme for Research and Training in Tropical Diseases Received 28 May
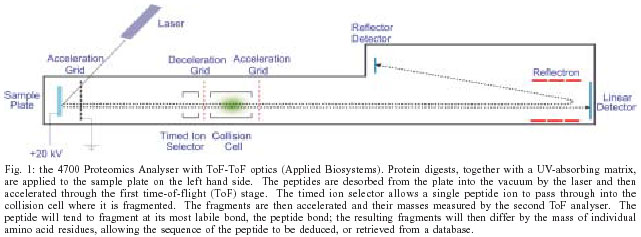
2004 Code Number: oc04085 An effective vaccine against schistosomiasis mansoni would be a valuable control tool and the high levels of protection elicited in rodents and primates by radiation-attenuated cercariae provide proof of principle. A major obstacle to vaccine development is the difficulty of identifying the antigens that mediate protection, not least because of the size of the genome at 280Mb DNA encoding 14,000 to 20,000 genes. The technologies collectively called proteomics, including 2D electrophoresis, liquid chromatography and mass spectrometry, now permit any protein to be identified provided there is extensive DNA data, and preferably a genome sequence. Applied to soluble (cytosolic) proteins from schistosomes, proteomics reveals the great similarity in composition between life cycle stages, with several WHO vaccine candidates amongst the most abundant constituents. The proteomic approach has been successfully applied to identify the secretions used by cercaria to penetrate host skin, the gut secretions of adult worms and the proteins exposed on the tegument surface. Soluble proteins can also be separated by 2D electrophoresis before western blotting to identify the full range of antigenic targets present in a parasite preparation. The next step is to discover which target proteins represent the weak points in the worm's defences. Key words: Schistosoma mansoni - vaccine - proteomics - mass spectrometry - antigen STRATEGIES FOR A HUMAN SCHISTOSOME VACCINE Most current viral and microbial vaccines were developed empirically, but in the knowledge that first exposure to the pathogen generated a strong immunity to re-infection. For parasites the situation is altogether more complex, not least because they have evolved efficient mechanisms to evade host immune responses. In the case of schistosomiasis mansoni, the result is a chronic debilitating infection that may persist for more than 30 years (Harris et al. 1984). In these circumstances the development of a schistosome vaccine was always going to be a difficult task. In what might be termed the classical approach, the strategy is to identify protected individuals in an endemic population. The immune mechanisms that such people deploy to limit or prevent establishment of invading cercariae should form the basis of a successful vaccine. In the last two decades great progress has been made in characterising human responses to schistosomes (Dunne & Mountford 2001) but no immune mechanisms or specific antigens strongly associated with a protected status have been identified. Indeed, in virtually all re-infection studies after curative chemotherapy, prepubertal children show little evidence of protection; these are the individuals with the highest intensities of infection who would benefit most from a vaccine. MODELS OF PROTECTION If the investigation of human responses to infection has not provided any obvious avenues to a vaccine, are there any model systems whose analysis might provide better pointers? The attenuated cercarial vaccine has long provided the gold standard for schistosome vaccine development. Its success in rodents and primates underlines the fact that protective immunity against schistosomes is a feasible prospect. The attenuated larvae must migrate a sufficient distance within the host to prime the immune system, without establishing a patent infection, and high levels of protection ensue (> 85% in baboons and mice; Mountford et al. 1996, Kariuki et al. 2004) It is important to stress that "protection" means reduction in worm burden, not proportion of hosts protected, as would be the case for microbial or viral vaccines. However, sterile immunity is probably not essential because a substantial reduction in worm burden would diminish both clinical disease and transmission. The immunity induced by the radiation-attenuated vaccine in mice has been the most thoroughly explored (Coulson 1997). A single exposure of C57BL/6 mice generates a predominantly cell-mediated protection, dependent on CD4+ T cells recruited to the lungs. These cells orchestrate a Th1-type focal inflammation against challenge larvae that effectively blocks their onward migration through the lung capillaries. We believe that the larvae trapped in the lung eventually expire, and certainly they do not succumb immediately to a lethal hit e.g. by nitric oxide from macrophages. With multiple exposures of mice to attenuated larvae, the antibody-mediated component of protection becomes more prominent. The timing of challenge elimination is again early after exposure, in either the skin or lungs, but neither the complement pathway nor Fc receptors seem to be essential (P Coulson, unpublished data), so the mechanism of antibody-mediated protection remains enigmatic. For primates, multiple exposures to attenuated cercariae are necessary to achieve a high level of protection and IgG levels at challenge provide the best correlation. There is no information on the site of parasite elimination or mechanism of protection but larvae again seem the likely targets. In addition to the attenuated vaccine, there are at least two other schistosome-host interactions that might form the basis for a vaccine, both involving hosts where the parasites establish but are then eliminated in a self-cure response. In the laboratory rat this occurs around four weeks after a primary infection when the worms are in the portal veins of the liver, coincident with rising IgE levels in the circulation and hepatic mast cell degranulation (Newlands et al. 1995, Cutts & Wilson 1997). Whilst such a mechanism might be difficult to replicate in a vaccine, the antigens that trigger the protective response should represent a chink in the parasite's armour. The rhesus monkey is an altogether more promising model where our recent observations suggest that parasite elimination, occurring between 12 and 18 weeks after infection, correlates with IgG level. As a result of immune pressure, the mature worms in the portal system first cease egg laying, then blood feeding and eventually starve to death (P Coulson, unpublished data). THE NEED TO IDENTIFY PROTECTIVE ANTIGENS If any of the above models is to serve as the basis for a vaccine, then it will be necessary to identify the relevant antigens, clone and express them as recombinant proteins (or insert them in a DNA vaccine vector) and formulate them for delivery to induce the desired immune response. Irrespective of the model, the proteins secreted by the parasite or expressed on its exposed epithelial surfaces are the obvious targets. (Glycan epitopes might also mediate protection but, whilst their characterisation is not an insuperable obstacle, would add a layer of complexity to the task of antigen identification.) For protein antigens the classical approach is to raise antisera against e.g. the secretions of cercariae or lung worms, and use these to screen cDNA expression libraries constructed with mRNA from the appropriate life cycle stage. However, when this approach was tried with the larval stages it yielded a very meagre harvest of novel antigens (Harrop et al. 1999, 2000). Instead, the sera detected the same panel of abundant, highly immunogenic, cytosolic or cytoskeletal antigens reported in other library screens; these incidentally included some of the vaccine candidates promoted by WHO (Bergquist et al. 2002). Our conclusion is that a radically new approach is needed to identify the antigenic targets of protection in any of the models described above. THE APPLICATION OF PROTEOMICS TO ANTIGEN IDENTIFICATION The proteome can be defined as the total protein complement of an organism, tissue, cell or subcellular organelle/complex. The technologies that have been developed over the last few years permit any protein to be identified, provided that an extensive cDNA or genomic database is available. The first step in the process is to separate the proteins in a complex mixture. For cytosolic or other soluble fractions this is readily achieved by 2D electrophoresis, with isoelectric focusing in the first dimension and SDS-PAGE in the second. Sensitive stains with a wide, linear dynamic range, such as Sypro Ruby (Molecular Probes, Oregon) make possible the visuali-sation of very small amounts of protein in the gel. The combination of a fluorescence imager to capture the gel image, and analysis software, allows spot patterns to be compared between gels to pinpoint differences in composition. Less soluble proteins, such as many cytoskeletal components, can also be made compatible with 2D electrophoresis by solubilisation in chaotropic agents and zwitterionic detergents (Molloy et al. 1998). After gel mapping, spots are excised and subjected to proteolytic digestion e.g. by trypsin that cuts peptide bonds C-terminal to lysine or arginine residues to create a diagnostic peptide mixture. After differential extraction the final fraction, containing integral membrane proteins, can only be solubilised in ionic detergents that are not compatible with isoelectric focusing; for such proteins a combination of 1D SDS-PAGE electrophoresis and/or liquid chromatographic (LC) separations must be used (Washburn et al. 2001). MASS SPECTROMETRIC IDENTIFICATION OF PROTEINS Mass spectrometry (MS) is now the method of choice for the identification of proteins (Ashton et al. 2001). It is possible, even with very small amounts of starting material, to confidently make a link between a protein spot on a gel and its encoding DNA sequence. For protein spots that have been excised from 2-D gels, the usual approach is to generate a peptide mass fingerprint (PMF). In theory, this should be diagnostic of a particular protein and searching a database of full-length protein sequences for the organism under investigation will lead to identification if the sequence for that protein is present in the database. However, with the increasing size of protein databases, it is now common to get false positive hits to e.g., very large and unrelated proteins. In analyses where some form of LC precedes the mass spectrometry, it is not even possible to produce a PMF, since the peptides from each of the proteins in the mixture will be separated into several different fractions. In both these cases, it is necessary to obtain identifications based not on the masses of the peptides from a single protein, but on the basis of individual peptides. This is achieved using tandem mass spectrometry. In a tandem mass spectrometer, there are two separate stages of mass analysis. The first stage allows a single peptide to be selected from a complex mixture. This peptide is then typically allowed to enter a collision cell containing an inert gas, where it is fragmented. The second stage then measures the masses of the fragments that are produced. Again this data can be used to search against a sequence database, but it is not necessary to have the full-length sequence of the protein under investigation since the technique is just as good at finding matches from EST data, or even from unassembled genomic sequences. All that is necessary is that some of the (partial) sequences in the database contain the region of the relevant gene, which encodes the peptide. Since the masses of the peptide fragments depend on the sequence of amino acids, this type of search is much more discriminating. In practice, identifications are usually based on the fragmentation patterns of several peptides, even if these are spread across multiple LC fractions. At York, we perform most of our mass spectrometry on a 4700 Proteomics Analyser (Fig. 1, Applied Biosystems). This instrument is capable of generating a peptide mass fingerprint and 10 peptide fragmentation spectra from a single protein in ~ 5 min, and can easily do this for up to 200 samples (or LC fractions) in a single run. THE SCHISTOSOME GENOME RESOURCE The S. mansoni genome contains 270 Mb of DNA encoding not less than 14,000 genes (Verjovski-Almeida et al. 2003), and perhaps as many as 20,000 (Franco et al. 1995). As of March 2004, it has been sequenced to > 9x coverage, but not yet assembled or annotated. However, the 2.9 million reads themselves provide a database against which MS spectra can be searched. Sequencing of the S. japonicum genome is also underway in China. A second and crucial resource, both for proteomics and gene finding is the expressed sequence tag (EST) database. Prior to 2003, approximately 14,000 ESTs had been deposited in the public domain, but a further 125,000 ESTs from six life cycle stages were added in October as a result of the efforts of a sequencing consortium in the state of São Paulo, Brasil, led by Dr Sergio Verjofski-Almeida, increasing the number of sequences that could be searched for a match by an order of magnitude (Verjovski-Almeida et al. 2003). Again, a Chinese project has produced ~ 45,000 ESTs from S. japonicum adult worms and eggs (Hu et al. 2003). It has been estimated that the São Paulo data has hits to ~ 92% of S. mansoni genes, but ~ 70% of the sequences have no homology to known proteins in other organisms, probably a reflection of the distinct phylogenetic position of schistosomes. Furthermore, it seems that on average around 7000 genes are expressed in each life cycle stage, about 1000 of which may be stage-specific; this information is very relevant for proteomic analysis. WHAT DOES PROTEOMICS REVEAL ABOUT SCHISTOSOME? A 2D separation of the most soluble proteins from any schistosome life cycle stage has two striking features. First, the preparation contains a complex mixture of several thousand spots (Fig. 2). Secondly, the spot pattern is remarkably similar, irrespective of the stage examined (Curwen et al. 2004). The greatest similarities were observed where the life-cycle stages were developmentally adjacent to one another. For example, lung schistosomula shared 90% of their analysed protein with adults. Eggs were the least similar to any other stage, probably as a result of their containing a free-living miracidium. Peptide mass fingerprinting of the 40 most abundant spots in soluble cercarial, lung worm, adult and egg extracts revealed that to a large extent, the dominant proteins were indeed shared. With the exception of one protein resident in the endoplasmic reticulum, all species identified by PMF were cytosolic in origin, as would be expected for a highly soluble cell fraction. Proteins with catalytic activity, particularly glycolytic enzymes, were dominant, along with several calcium binding proteins. Actin and other muscle components were also abundant, understandable given the predominance of this tissue throughout all life-cycle stages. The majority of these abundant proteins were originally identified by virtue of their immunological properties, either as antibody targets in previously infected hosts or as putative vaccine candidates. Why should the different stages show such a similar composition and what are the implications? Schistosomes possess tissues differentiated into rudimentary organs such as gut, nervous system, musculature and excretory system. However, as members of the Phylum Platyhelminthes, they have a solid body plan from which these individual organs cannot be dissected out. Simple extraction of the whole parasite body therefore averages for composition across the various tissues so that constituents common to all, such as glycolytic enzymes, are dominant. It is also striking that several of the vaccine candidates promoted by WHO are dominant within the cytosolic and cytoskeletal fractions. Given our stated objective of identifying secreted or surface-exposed proteins as potential vaccine candidates, how do we circumvent the problems of complexity and similarity in the parasite fractions? The solution is targeted proteomics; the parasite samples to be characterised need a simpler composition. This can be achieved e.g., by collecting the secretions released from a specific larval stage. However the strategy carries with it the cost of much lower protein yields, but hopefully the material obtained will be of much greater relevance to vaccine development. With these caveats in mind we have developed an in vitro scheme for analysing the proteins released by the cercaria as it transforms to the schistosomulum, and by the larvae in culture up to 8-10 days, by which time they are fully developed to the lung stage. The cercarial secretions are the most complex, in part due to the holocrine nature of secretion from the acetabular glands. (The entire contents of the gland are squeezed out, so that both cytosolic and vesicular proteins are present.) Both cercarial elastase (Newport et al. 1988) and Sm16 (Rao & Ramaswamy 2000), two previously characterised cercarial proteins, are dominant in the preparation, plus a number of other novel proteins. These, together with the molecules secreted by the skin and lung stage larvae (~ 20 in total) will, when fully characterised, cloned and expressed, form a panel of new antigens to be tested in vaccination experiments designed to emulate the attenuated cercarial vaccine. We are developing a similar approach to characterise the gut secretions of adult worms, where at present only 2-3 proteases have been identified. Apart from the parasite eggs, these gut secretions represent the major source of material released by the adults into the host bloodstream and hence a likely source of antigens relevant to the rhesus and rat self-cure models. The only other major source of antigens that could serve as vaccine candidates is the surface tegument of both larvae and adults. This syncytial layer, which envelops the entire worm surface, is bounded by a normal plasma membrane overlain by an apparently inert and lipophilic secreted bilayer, or membranocalyx, which provides a barrier against immune attack. The plasma membrane has documented transport functions and enzyme activities, and the exposed portions of the proteins involved may represent vulnerable points for attack, especially relevant to the rhesus model. It is also unclear whether the membranocalyx contains proteins that could serve as immune targets. The proteomic approach will allow the molecular architecture of the parasite surface to be established, defining both the extent of possibilities for immune attack and the nature of the very successful immune evasion mechanism. THE PARASITE GLYCOME It is worth mentioning that the mass spectrometric approach can also be applied to characterise the glycan moieties of glycoproteins and glycolipids that can function as antigenic epitopes, provoking antibody production. Given the potent response that primates make to such glycans, we have questioned whether this reactivity represents a smokescreen rather than a component of protection (Eberl et al. 2001). This is especially true of the parasite egg that seems to advertise its presence by releasing proinflammatory secretions to provoke a response essential for its transit through tissues to the gut lumen (Doenhoff et al. 1986). The glycan residues must first be released from the parent protein(s) by PNGase F treatment for N glycans and PNGase A treatment, plus reductive elimination, for the O glycans, prior to mass spectrometry. Empirical structures can be predicted from the mass spectra obtained but structure assignments then need to be confirmed by further rounds of exo- and endo-glycosidase digestions, and MS. Our preliminary studies in collaboration with Professor Anne Dell of Imperial College, London on the glycoproteins released by cercariae during skin penetration, and mature eggs in culture, reveal a rich mixture of both N and O glycans. Moreover, the majority of cercarial N glycans appear to be shared with eggs, but the latter contain a lot of glycans structures not present in the cercarial secretions. The fact that immunisation with eggs fails to elicit protection is further evidence that the cross-reactive N glycans are a smok-escreen. Assigning definitive structures and biological functions to the various glycans will be a major task. THE IMMUNOME The immunome can be defined as that subset of the proteome that reacts with the immune system, most easily demonstrated where proteins act as targets for antibody. Clearly, in vaccine development, where protection is demonstrably mediated by antibodies it is desirable to pinpoint the precise proteins and even epitopes that mediate binding. An important caveat is that the presence of an antigenic epitope on a protein does not make it protective. Indeed, most reactivities will be spurious clues, and the stringent use of non-protective control sera is essential to eliminate them. Western blotting is the method of choice to identify reactive proteins. It is necessary to run duplicate 2D gels of a protein preparation, one of which is then blotted onto a PVDF membrane before gel and blot are stained with Sypro Ruby (using different protocols) hopefully to reveal identical patterns of proteins; the blot is then probed to visualise the targets. Careful matching of blot to gel pinpoints the protein subset to be excised, digested and subjected to MS to obtain identities. (N.B. It is not possible to get protein identities directly by excision of blot spots because the presence of the two antibodies, the enzyme label and the blocking protein swamps other signals.) In the context of schistosomiasis we are currently exploring the reactivity of sera from mice given multiple exposures of the attenuated cercarial vaccine, and rhesus monkeys that have undergone self-cure. Not only have the conventional soluble antigen preparations from different life cycle stages (SCAP, SLAP, SWAP, SEA) proved very similar in composition (Curwen et al. 2004) but their cytosolic (GST, Sm14) or cytoskeletal (paramyosin) components promoted as vaccine candidates are also the most immunoreactive proteins. The rational explanation is that such proteins possess immunogenicity because they have not been subject to immunological pressure during evolution due to their internal location. Consequently the ~ 20% protection often reported after vaccination with such antigens may be their maximum potential. Our principal task is to pinpoint the reactive antigens within the preparations highly enriched in secretory and membrane proteins that we are developing, since these should include the proteins that represent the parasite's Achilles heel. We also need to consider the contribution made to reactivity by glycan epitopes, especially in view of the degree of glycan-sharing between proteins, which results in cross-reactivity. With respect to the immunome, there are two technical problems that still need to be solved before the full power of the approach can be realised. The inability to separate membrane proteins by 2D electrophoresis is one limitation. The only way in which such proteins can be blotted is after a 1D separation, which provides inadequate resolution of the mixture, such that a reactive band may contain one to several proteins and it will be unclear which is the real antibody target. The second is the difficult task of screening a protein preparation for T cell reactivity at the level of individual constituents that can be identified by MS. CONCLUSIONS It has been possible for 20 years to perform 2D separations of complex protein mixtures, but only recently have technical developments made such gels reproducible. The parallel developments in the mass spectrometry of proteins have also made it possible to identify the many proteins present, provided the DNA sequence data exists. Thus the scene is set, given adequate funding, to identify the complete subset of secreted and surface-exposed schistosome proteins from which the possible vaccine candidates will emerge. In our estimation the targeted proteomic approach will reduce the potential candidates from the not less than 14,000 proteins encoded by the genome, to a maximum of 100-200 dominant proteins in the secretory and membrane preparations. This is a much more manageable number, and the information should be available within in a time frame of 2-3 years. ACKNOWLEDGEMENTS To Mrs Ann Bamford and Ms Shobana Sundaralingam for expert technical assistance REFERENCES
Copyright 2004 Instituto Oswaldo Cruz - Fiocruz. The following images related to this document are available:Photo images[oc04085f1.jpg] [oc04085f2.jpg] |
| |||||||||

{kind=link}
{kind=link}