 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol.100, Suppl. 1, March, 2005, pp. 19-23 Nitric Oxide paradox in asthma Alexandre Castro Keller, Dunia Rodriguez, Momtchilo Russo+ Departamento de Imunologia, Instituto de Ciências Biomédicas, Universidade de São Paulo, 05508-900 São
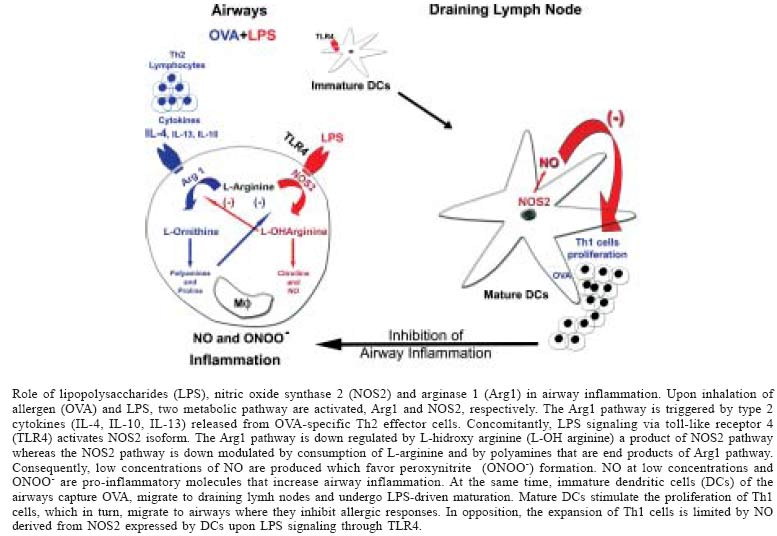
Paulo, SP, Brasil Received 8 November 2004 Code number: oc05022 Asthma results from allergen-driven intrapulmonary Th2 response, and is characterized by intermittent airway obstruction, airway hyperreactivity (AHR), and airway inflammation. Accumulating evidence indicates that inflammatory diseases of the respiratory tract are commonly associated with elevated production of nitric oxide (NO). It has been shown that exhaled NO may be derived from constitutive NO synthase (NOS) such as endothelial (NOS 3) and neural (NOS 1) in normal airways, while increased levels of NO in asthma appear to be derived from inducible NOS2 expressed in the inflamed airways. Nevertheless, the functional role of NO and NOS isoforms in the regulation of AHR and airway inflammation in human or experimental models of asthma is still highly controversial. In the present commentary we will discuss the role of lipopolysaccharides contamination of allergens as key element in the controversy related to the regulation of NOS2 activity in experimental asthma. Key words: asthma - nitric oxide - lipopolysaccharides Allergic asthma is a chronic respiratory disease characterized by allergen-induced early and late bronchial obstructive reactions that are associated with IgE antibodies, mast cells and infiltration of T helper 2 (Th2) cells, eosinophils, and neutrophils in the airways. These cells secrete type 2 cytokines and inflammatory mediators that lead to the development of airway hyperresponsiveness (AHR) to a variety of stimuli, including allergens, chemical irritants, and pharmacological agents such as histamine and methacholine (Bousquet et al. 2000). As a result of the chronic inflammatory process, the airways may suffer profound structural changes that include epithelial metaplasia, fibrosis and increased airway smooth muscle mass, referred to as airway remodeling (Elias et al. 1999, Bousquet et al. 2000). Nitric oxide (NO) is now well recognized for its involvement in diverse biological processes, including vasodilation, bronchodilation, and regulation of inflammatory-immune processes. Thus, it is not surprising that the role NO in asthma has been under investigation. Accumulating evidence indicate that NO plays a role in the regulation of airway function both in health and disease. Indeed, exhaled NO has been detected in normal subjects and asthmatics (Barnes & Liew 1995, Saleh et al. 1998, Ricciardolo 2003). Exhaled NO could be generated enzymatically by three distinct isoforms of NO synthase (NOS1, NOS2, NOS3) expressed by different cell types present in normal or inflamed lung tissue such as airway bronchial epithelial cells, neuronal cells, macrophages, eosinophils, neutrophils, mast cells, and endothelial and smooth muscle cells. NOS1 and NOS3 isoforms are expressed constitutively and produce low levels of NO whereas NOS2 isoform is induced by bacterial products and/or pro-inflammatory cytokines (Meurs et al. 2003). As such, in asthmatics or other inflammatory lung diseases the exhaled NO appears to derive from NOS2 expressed within bronchial epithelial cells or immuno-inflammatory cells (Kharitonov et al. 1994, Barnes 1995, Guo et al. 2000, Van Der Vliet et al. 2000, Yates 2001). Indeed, the level of NO concentration in exhaled air is considered to be a marker of airway inflammation and the exhaled NO observed in asthmatics has been associated with airway eosinophilia and AHR (Saleh et al. 1998, Ricciardolo 2003). Thus, it appears that NO has a harmful and pro-inflammatory role in asthma. However, there is no clear clinical evidence that long-term inhibition of NOS2 activity could have a therapeutic value in patients with asthma. This issue is further unsettled because several groups that investigated the role of NOS2 in the murine asthma model generated conflicting data. For instance, it has been shown that acute inhibition of NOS2 activity has no effect (Feder et al. 1997), suppresses (Trifilieff et al. 2000), or exacerbates airway inflammation (Blease et al. 2000). Controversial data was also generated in studies using NOS2 knockout mice. Xiong et al. (1999), showed that NOS2-/- animals presented a diminished airway inflammation while others clearly showed that airways inflammation is fully expressed in NOS2-/- mice (De Sanctis et al. 1999, Rodriguez et al. 2003). We will comment here some of the conflicting reports regarding the participation of NO and particularly NOS2 in experimental asthma. We would put forward a hypothesis that contamination of allergens with lipopolysaccharides (LPS) might be at the center of controversy regarding the participation of NOS2 in asthma. NOS2 INHIBITORS AND NOS2-DEFICIENCY IN EXPERIMENTAL ASTHMA To assess the contribution of NOS isoforms to airway eosinophilic inflammation AHR, studies were performed with selective NOS inhibitors or with animals with targeted deletions of the three NOS isoforms. Feder et al. (1997) investigated the effect of four NOS inhibitors, N-nitro-L-arginine methyl ester (L-NAME), aminoguanidine, NG-onomethyl-L-arginine (NMMA), and L-N6-(1-Iminoethyl) lysine (L-NIL), on the influx of eosinophils into the bronchoalveolar lavage (BAL) fluid and lung tissue of sensitized and OVA-challenged mice. The authors found that treating the animals with L-NAME, amino-guanidine or NMMA significantly reduced the OVA-induced pulmonary eosinophilia. In contrast, treatment with L-NIL, a selective NOS2 inhibitor, had no effect on airway eosinophilia. In addition, the expression of NOS2 mRNA or NOS2 protein was not increased in the lungs of OVA-challenged mice when compared to control mice. Also nitrite levels measured in BAL fluid were not increased. The authors conclude from these results that NO contribute to OVA-induced eosinophilia but its production is not generated through the activity of inducible NOS2 (Feder et al. 1997). In contrast, Trifilieff et al. (2000) using a similar approach, found that two different selective inhibitors of NOS2, EIT (S-ethylisothiourea) or AMT (2-amino-5,6-dihydro-6-methyl-4H-1,3-thiazine), administered during the challenge period, markedly reduced AHR and BAL eosinophilia. In addition, the authors found that allergic mice presented an increased NOS activity that was mainly calcium-independent, suggesting the involvement of NOS2. Moreover, lung sections of sensitized and OVA-challenged mice were positive for NOS2 immunostaining whereas lung sections from PBS-challenged mice showed no positive staining. Positive NOS2 staining was present mainly in the cells among the inflammatory infiltrate. Finally, in another set of experiments the authors showed that NOS2 promotes airway lung inflammation via direct up-regulation of chemokine expression (Trifilieff et al. 2000). These two studies illustrate well the controversy regarding data generated with NOS inhibitors. Strikingly, studies in mice with targeted deletions of the three isoforms of NOS also generated conflicting results. For instance, De Sanctis et al. (1999) compared the asthma-like response of wild-type (WT) animals with those obtained in mice with NOS-deficiencies. The authors found that total NOS activity increased in sensitized and OVA-challenged WT animals as well as in NOS1 and 3 double KO animals, but not in NOS2-KO mice. These results indicate that the enhanced NOS activity detected in the lungs of allergic mice is dependent of NOS2 expression. However, no significant differences were observed in AHR and eosinophlic inflammation when comparing WT animals with NOS2-deficient mice. These results clearly indicate that NOS2 activity is increased in the airways of allergic mice, but NOS2-deficiency did not affect the asthma phenotype. In contrast, Xiong et al. (1999) showed that infiltration of inflammatory eosinophils, loss of structural integrity of the airway walls, microvascular leakage, pulmonary edema, and airway occlusion but not AHR were markedly less severe in the NOS2 mutants than in WT animals. Surprisingly, all studies used similar protocols for sensitization and allergen challenge. Can these apparently discordant findings be reconciled? Before addressing this issue, it will be of importance to ask why NOS2 should be up regulated in experimental asthma? On the basis of current knowledge, it is conceivable that NOS2 expression or NO production in allergic lungs will be suppressed because type 2 cytokines including IL-4, IL-13, and IL-10 induce the expression of Arginase 1 (Arg 1) (Modolell et al. 1995, Hesse et al. 2001). The Arg1 metabolic pathway depletes L-arginine and consequently limits NO synthesis by NOS2 (Wu & Morris 1998). Actually, the final products of Arg 1 pathway are the formation of polyamines and proline that are respectively involved in cell proliferation and collagen formation, which are key features of airway remodeling (Elias et al. 1999, Zim-mermann et al. 2003). Moreover, polyamines down regulate NOS2 activity (Bronte et al. 2003). As such, Arg1 but not NOS2 should be up regulated in asthma. One possibility for NOS2 expression in experimental asthma is the presence of endotoxin LPS, a cell wall component of gram-negative bacteria, which signals through toll-like receptor 4 (TLR4) and leads to up regulation of NOS2 activity (Wagner et al. 1983, Stuehr & Marletta 1985). It is known that LPS are ubiquitous in the environment and are found in household dusts allergens and allergen extracts (Michel et al. 1991, Gereda et al. 2000), and are common contaminant of commercial OVA (Watanabe et al. 2003). Interestingly, it was recently reported that low level of inhaled LPS is necessary to induce Th2 responses to inhaled OVA in a mouse model of allergic sensitization that do not use adjuvant for sensitization (Eisenbarth et al. 2002). Thus, in addition to NOS2 induction, LPS appears to be involved in eosinophils recruitment. In contrast, high doses of LPS appear to suppress eosinophilic inflammation and type 2 cytokine production, however high LPS doses promote neutrophil recruitment and Th1 responses (Eisenbarth et al. 2002, Rodriguez et al. 2003). Considering LPS as a common contaminant of OVA or house dust allergens we envisage the following scenario: after OVA (allergen) and LPS inhalation, two enzymatic pathways, Arg1 and NOS2, will be respectively activated. The Arg1 pathway is activated by type 2 cytokines released from allergen-driven Th2 cells whereas LPS activate NOS2 pathway (see Figure). It is known that L-OH Arginine, a byproduct of the conversion of L-Arginine by NOS2, inhibits Arg1, whereas Arg1 depletes L-arginine and generates polyamines that limits NO production and down regulate NOS2 expression, respectively (Bronte et al. 2003, Meurs et al. 2003). Since both pathways are activated and are reciprocally down-regulated, it is likely that L-Arginine concentrations and NO production are at low levels. It has been shown the NOS2-dependent catabolism of L-Arginine at low levels favor the production of peroxinitrite (ONOO-), a potent pro-inflammatory molecule (Xia & Zweier 1997, Xia et al. 1998). Moreover, low local levels of NO promote the expression of pro-inflammatory proteins including COX2 and IL-6 (Connelly et al. 2001). Another aspect related to LPS contamination is the fact that LPS through TLR4 signaling drives the maturation of dendritic cells (DCs). Thus, immature DCs from the airways upon LPS stimulation migrate to regional lymph nodes where they mature and expand allergen-specific T cells that produce IFN-g (Netea et al. 2004). In turn, these Th1 cells might migrate to airways and could dampen allergic Th2 responses. Conversely, LPS-induced NOS2 activity in DCs, results in the production of NO and/or peroxinitrite that are known to inhibit Th1 cell expansion by apoptosis (see Figure). In summary, NOS2 activity induced by LPS contamination of allergens might affect asthma development in two ways: inducing proinflammatory cytokines in the airways and suppressing Th1 expansion in regional lymph nodes. Indeed Xiong et al. (1999) showed that enhanced of IFN-g production in NOS2-deficient mice was apparently responsible for the suppression of both eosinophilia and disease, as in vivo depletion of IFN-g restored allergic pathology in these animals. We speculate that in this case, LPS contamination was responsible for the emergence of OVA-specific Th1 cells. In the same vein, it is possible that in experiments where inhibition of NOS2 ameliorated airway inflammation, LPS could have contaminated the allergen (Trifilieff et al. 2000). It should be pointed out that high concentration of LPS appear to suppress asthma-like responses. LPS acts at two levels; it suppresses the emergence of allergen-driven Th2 cells (Eisenbarth et al. 2002) and as we have recently shown, LPS signaling through TLR4 suppresses established airway eosinophilic inflammation via NOS2 activity (Rodriguez et al. 2003). It is known that either microbial infections, or local administration of type 1 cytokines IL-12 and IFN-g appear to prevent allergic responses (de Sousa Mucida et al. 2003). These agents are also involved in the induction of NO production. We have previously examined the effect of systemic endotoxin (LPS) administration on established airway inflammation and AHR in a murine asthma model. In our protocol, we gave two OVA challenges at weekly intervals because, 24 h after the second OVA challenge the asthma-like responses including airway inflammation, airway hyperactivity, mucus, and cytokine production are synchronized. Moreover, after the first OVA challenge the airway inflammation is initiated allowing us to determine the effect of LPS on established airway inflammation. The LPS administration was given intravenously concomitantly with the second OVA i.n. challenge. We found that systemic LPS administration suppressed multiple effector responses of asthma. The suppressive activity of LPS on airway inflammation, and on mucus and type 2 cytokine production persisted in IL-12 or IFN-g-deficient mice. However, in mice lacking NOS2, the suppressive effect of LPS was absent. These results support the notion that LPS administration suppresses the allergic eosinophilic inflammation and hyperresponsiveness through a NO-dependent mechanism generated via NOS2. These data clearly indicate that LPS-induced NOS2 activation result in a powerful anti-allergic activity. In summary, we have highlighted the complexity of NOS2 biochemistry in asthma and discussed that NOS2 activation could have both pro- and anti-inflammatory effects. It is possible that these effects could be critical- Finally, it is still unclear whether NOS2 inhibition would be a good therapeutic target in asthma because it might favor the activation of Arg1 pathway that is involved in airway remodeling. We suggest that the detrimental effect of NOS2 in asthma is related to the L-arginine and NO concentrations because NO at high concentration has a clear anti-inflammatory effect (Honda et al. 1999, Connelly et al. 2001, Rodriguez et al. 2003). Thus, inhalation of NO could be a potential therapeutic strategy to suppress lung inflammation. REFERENCES
Copyright 2005 Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc05022f1.jpg] |
| |||||||||

{kind=link}