 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
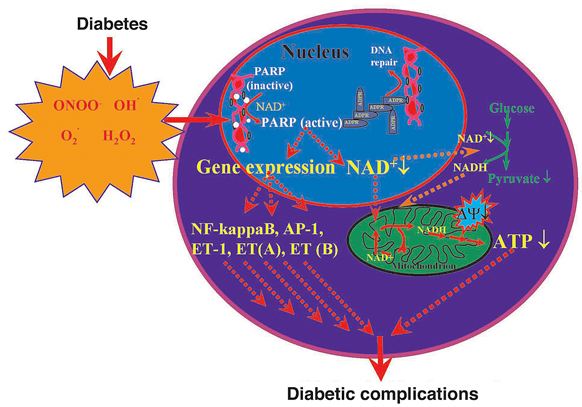
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol.100, Suppl. 1, March, 2005, pp. 29-37 The pathogenesis of diabetic complications: the role of DNA injury and poly(ADP-ribose) polymerase activation in peroxynitrite-mediated cytotoxicity Levente Kiss, Csaba Szabó/*/+ Institute of Human Physiology and Clinical Experimental Research, Semmelweis University, Budapest, Hungary *Inotek Pharmaceuticals, Suite 419E,100 Cummings Center, Beverly, MA 01915, US Financial support: NIH, Hungarian Research Fund Received 8 November 2004 Code number: oc05024 Recent work has demonstrated that hyperglycemia-induced overproduction of superoxide by the mitochondrial electron-transport chain triggers several pathways of injury [(protein kinase C (PKC), hexosamine and polyol pathway fluxes, advanced glycation end product formation (AGE)] involved in the pathogenesis of diabetic complications by inhibiting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity. Increased oxidative and nitrosative stress activates the nuclear enzyme, poly(ADP-ribose) polymerase-1 (PARP). PARP activation, on one hand, depletes its substrate, NAD+, slowing the rate of glycolysis, electron transport and ATP formation. On the other hand, PARP activation results in inhibition of GAPDH by poly-ADP-ribosylation. These processes result in acute endothelial dysfunction in diabetic blood vessels, which importantly contributes to the development of various diabetic complications. Accordingly, hyperglycemia-induced activation of PKC and AGE formation are prevented by inhibition of PARP activity. Furthermore, inhibition of PARP protects against diabetic cardiovascular dysfunction in rodent models of cardiomyopathy, nephropathy, neuropathy, and retinopathy. PARP activation is also present in microvasculature of human diabetic subjects. The present review focuses on the role of PARP in diabetic complications and emphasizes the therapeutic potential of PARP inhibition in the prevention or reversal of diabetic complications. Key words: poly(ADP-ribose) polymerase - peroxynitrite - nitric oxide - diabetic complications - oxidative stress - endothelial dysfunction POLY(ADP-RIBOSE) POLYMERASE AND ITS ACTIVATION Poly(ADP-ribose) polymerase (PARP) is an abundant nuclear enzyme with complex regulatory functions (Hiromatsu et al. 1992, De Murcia et al. 1994, Menissier-de Murcia et al. 1997, Szabó et al. 1997b, 1998a, 2000, Le Rhun et al. 1998, Rudat et al. 1998, De Murcia &Shall 2000, Davidovic et al. 2001, Virág & Szabó 2002). Poly(ADP-ribose) polymerase-1 (PARP-1, EC 2.4.2.30) [also known as poly(ADP-ribose) synthetase (PARS)], the major PARP isoform, is a member of the PARP enzyme family consisting of PARP-1 and many additional, recently identified poly(ADP-ribosylating) enzymes (minor PARP isoforms). Because the role of the minor PARP isoforms is poorly understood, in this review we alternate the use `PARP-1' and `PARP'. Numerous overviews and monographs appeared in recent years about the structure and functions of PARP (Szabó et al. 1998a, 2000, Virág & Szabó 2002, Zhang 2002). PARP is a DNA damage sensor and signaling molecule and binds to both single- and double stranded DNA breaks, becomes activated, forms homodimers and initiates an energy-consuming cycle by transferring ADP ribose units from NAD+ onto nuclear acceptor proteins such as histones, transcriptional factors, DNA replication factors and signaling molecules (AP-1, DNA-PK, NFkB, Oct-1, p53) with PARP itself being the major acceptor. Auto-poly(ADP-ribosyl)ation represents a major regulatory mechanism for PARP resulting in the down-regulation of the enzyme activity. Poly(ADP-ribosyl)ation alters the activity of the other acceptor proteins also, due to the high negative charge of the attached ADP-ribose. In case of the histones, this process leads to electrostatic repulsion between DNA and loosening up the chromatin structure thereby making genes more accessible for gene transcription (Oikawa et al. 1980, Poirier et al. 1982, Park et al. 1983, Satoh & Lindahl 1992, Lautier et al. 1993, Herceg et al. 2001). The effect of PARP on the function of these proteins is carried out by non-covalent protein-protein interactions and by covalent poly(ADP-ribosylation) (for review see Virág & Szabó 2002). Poly(ADP-ribosylation) is a dynamic process as the polymer is rapidly degraded by two enzymes: poly(ADP-ribose) glycohydrolase (PARG) and ADP-ribosyl protein lyase. These enzymes are involved in the catabolism of poly(ADP-ribose) with PARG cleaving ribose-ribose bonds of both linear and branched portions of poly(ADP-ribose) and the lyase removing the protein proximal ADP-ribose monomer (Davidovic et al. 2001). The biological role of PARP is complex. One of the most important functions is related to the fact that mild genotoxic noxa cause PARP activation that facilitates DNA repair and cell survival. Under pathophysiological conditions, reactive species (such as hydrogen peroxide, hydroxyl radical, and peroxynitrite) trigger severe DNA damage and overactivation of PARP (Szabó et al. 1996, 1998b). Peroxynitrite is considered a key trigger of DNA strand breakage because it can travel significant distances and readily crosses cell membranes. When activated by DNA breaks, PARP initiates an energy-consuming cycle resulting in the depletion of NAD+ and ATP, slowing the rate of glycolysis and mitochondrial respiration, eventually leading to cellular dysfunction and necrotic cell death. Necrosis is the least desirable form of cell death as the cellular content is released into the tissue exposing neighboring cells to proteases and various pro-inflam-matory factors, thereby triggering positive feedback pathways of inflammatory tissue injury (Virág & Szabó 2002). PARP-overactivation induced necrosis is also related to intracellular acidification (Herceg et al. 2001, Affar et al. 2002). Another important function of the PARP enzyme is the regulation of the production of inflammatory mediators such as inducible nitric oxide synthase (iNOS), intercel-lular adhesion molecule-1 (ICAM-1) and major histocompatibility complex class II (MHC Class II) (Hiromatsu et al. 1992, Ehrlich et al. 1995, Szabó et al. 1997b, Zingarelli et al. 1998, Simbulan-Rosenthal et al. 2000 ). NF-kB is a key transcription factor in the regulation of this set of proteins and PARP has been shown can act as a co-activator in the NF-kB-mediated transcription. Other roles of PARP include its involvement in the regulation of replication and differentiation. It has been shown that poly(ADP-ribose) polymer can also serve as an emergency source of energy used by the base excision machinery to synthesize ATP (Oei et al. 2000). Furthermore, poly(ADP-ribose) may also serve as a signal for protein degradation in oxidatively injured cells (Ullrich et al. 2000). It must be stressed that PARP functions as a double-edged sword: on one hand, moderate activation of PARP can be of physiological importance via enhancement of the DNA repair. On the other hand, overactivation of PARP represents an important mechanism of tissue damage in various pathological conditions associated with oxidative and nitrosative stress, including myocardial reperfusion injury (Zingarelli et al. 1998), heart transplantation (Szabó et al. 2002), heart failure (Pacher et al. 2002b, c), stroke (Eliasson et al. 1997, Komjáti et al. 2004), circulatory shock (Szabó et al. 1997a, Oliver et al. 1999, Jagtap et al. 2002, Soriano et al. 2002, Pacher et al. 2002a, Shimoda et al. 2003), and autoimmune beta-cell destruction associated with diabetes mellitus (Burkart et al. 1999, Pieper et al. 1999). Activation of PARP and beneficial effects of various PARP inhibitors have been demonstrated in various forms of endothelial dysfunction such as the one associated with circulatory shock, hypertension, atherosclerosis, preeclampsia and aging (Szabó et al. 1997, Hung et al. 2002, Martinet et al. 2002, Pacher et al. 2002e, f). Furthermore, recent data suggest that activation of PARP importantly contributes to the development of endothelial dysfunction in various experimental models of diabetes and also in humans (Garcia Soriano et al. 2001, Soriano et al. 2001a, Pacher et al. 2002d, Szabó et al. 2002b). In addition, it has recently been demonstrated that PARP activation plays a pathogenetic role in diabetic nephropathy, neuropathy and retinopathy (Szabó et al. 2001, Minchenko et al. 2003, Li et al. 2004). PARP IN THE PATHOGENESIS OF DIABETIC ENDOTHELIAL DYSFUNCTION In established diabetic patients the quality of life and life expectations are determined by the complications of the disease. Endothelial dysfunction is a well documented complication in various forms of diabetes, and even in pre-diabetic individuals (Ruderman et al. 1992, Cosentino & Luscher 1998, Caballero et al. 1999, Cai & Harrison 2000, Calles-Escandon & Cipolla 2001, Szabó et al. 2002b). The pathogenesis of this endothelial dysfunction includes increased polyol pathway flux, altered cellular redox state, increased formation of diacylglycerol, activation of specific protein kinase C isoforms, and accelerated nonenzymatic formation of advanced glycation endproducts. Many of these pathways trigger the production of oxygen- and nitrogen-derived oxidants and free radicals, such as superoxide anion and peroxynitrite, which play a significant role in the pathogenesis of the diabetes-associated endothelial dysfunction and other diabetic complications. The cellular sources of reactive oxygen species such as superoxide anion are multiple and include advanced glycation endproducts, NADH/NADPH oxidases, the mitochondrial respiratory chain, xanthine oxidase, the arachidonic acid cascade, and microsomal enzymes (Giugliano et al. 1996, De Vriese et al. 2000, Nishikawa et al. 2000, Brownlee 2001, Beckman 2002, Guzik et al. 2002, Ceriello 2003b). In murine and human endothelial cells, the oxidative and nitrosative stress induced by elevated glucose causes DNA-damage and subsequent PARP activation (Garcia Soriano et al. 2001). The role of hyperglycemia-induced oxidative stress in producing DNA damage is also supported by recent findings showing that increased amounts of 8-hydroxyguanine and 8-hydroxydeoxy guanosine (markers of oxidative damage to DNA) can be found in both the plasma and tissues of streptozotocin diabetic rats (Park et al. 2001). Base modifications and DNA strand breaks have been demonstrated in diabetic patients as well (Lorenzi et al. 1987, Anderson et al. 1998, Astley et al. 1999, Sardas et al. 2001, Dincer et al. 2003). In other experiments, we have shown that the diabetes-associated loss of endothelial function is not only preventable, but also rapidly reversible with PARP inhibition. Intravascular PARP activation in endothelial cells and in vascular smooth muscle cells was already apparent 2 weeks after the onset of diabetes and thus it preceded the occurrence of the endothelial dysfunction, which developed between the 2nd and the 4th week of diabetes. Starting the treatment with the PARP inhibitor 1 week after streptozotocin ameliorated vascular poly(ADP-ribose) accumulation and restored normal vascular function without altering systemic glucose levels, plasma glycated hemoglobin levels, or pancreatic insulin content and restored the already established diabetic endothelial dysfunction (Garcia Soriano et al. 2001, Soriano et al. 2001a). Furthermore, even in vitro incubation of diabetic blood vessels with PARP inhibitors significantly enhanced their endothelium-dependent relaxant responsiveness (Soriano et al. 2001a). The potential of PARP inhibition in reversing endothelial dysfunction has also been demonstrated in an autoimmune model of diabetes (Pacher et al. 2002d). The development of the endothelial dysfunction was associated with a simultaneous loss of NAD+ and NADPH in the vasculature, and PARP inhibition reversed these changes. Based on these observations, and the known fact that eNOS is dependent on NADPH and is sensitively regulated by this co-factor, we proposed that the endothelial dysfunction in diabetes is dependent on a PARP-mediated, reversible cellular NADPH deficiency (Garcia Soriano et al. 2001, Soriano et al. 2001a). This proposal is also supported by prior work in oxidatively challenged cells in vitro (Hoyt et al. 1994, Landmesser et al. 2000, Cole et al. 2002). It is interesting to note, that other groups have demonstrated that diabetic endothelial dysfunction is also associated with direct oxidation and consequent cellular depletion of other co-factors of eNOS, such as tetrahydrobiopterin (BH4) (Guzik et al. 2000, Garcia Soriano et al. 2001, Fukuda et al. 2002, Pannirselvam et al. 2002, Werner-Felmayer et al. 2002) leading to increased free radical and oxidant production, oxidative damage and further exacerbation of the endothelial dysfunction (Virág & Szabó 2002). Two mechanisms appear to be involved in the protective action of PARP inhibition on the vascular endothelium in vivo: the conservation of cellular energetic pools and the prevention of the up-regulation of various pro-inflammatory pathways (cytokines, adhesion molecules, mononuclear cell infiltration) triggered by hyperglycemia (Garcia Soriano et al. 2001, Soriano et al. 2001a, Ceriello et al. 2003a). Recent data indicate that pharmacological inhibition of PARP can suppress the latter process suppressing the NF-kappaB activation and the expression of adhesion molecules both under constant high glucose as well as under intermittent high/low glucose conditions in cultured endothelial cells in vitro (Ceriello et al. 2003a). Intermittent high/low glucose induced a more pronounced expression of adhesion molecules than constant high glucose, and this process is regulated by PARP (Quagliaro et al. 2003, Piconi et al. 2004). Recent studies have also demonstrated that the hyperglycemia-induced overproduction of superoxide by mitochondrial electron-transport chain activated major pathways of hyperglycemic damage found in aortic endothelial cells [activation of protein kinase C (PKC) isoforms, hexosamine pathway flux, and advanced glycation end product formation (AGE)] by inhibiting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) activity (Du et al. 2003, Reusch 2003). GAPDH inhibition was found to be a consequence of poly(ADP-ribosyl)ation of GAPDH by PARP, which was activated by DNA strand breaks produced by reactive species, most likely peroxynitrite, generated by hyperglycemia. Both the hyperglycemia-induced decrease in activation of GAPDH and its poly(ADP-ribosyl)ation can be prevented by overexpression of either uncoupling protein-1 (UCP-1) or manganese superoxide dismutase (MnSOD), which decrease hyperglycemia-induced superoxide generation (Du et al. 2003). Similarly, administration of the mito-chondrial uncoupler 2,4-dinitrophenol (DNP) to endothelial cells exposed to high glucose blocked the glucose induced DNA strand breakage and PARP activation (Zsengellér et al. 2004). Importantly, the hyperglycemia-induced activation of PKC isoforms, hexosaminase pathway flux and AGE formation were prevented by the pharmacological blockade or genetic inactivation of PARP-1 activity (Du et al. 2003). Another factor to be considered in the process of diabetic endothelial dysfunction is angiotensin II, which is known to play a role in the pathogenesis of diabetic complications, perhaps most importantly in nephropathy, cardiomyopathy and retinopathy. Recent studies indicate that the protective effects of angiotensin converting enzyme inhibitors or angiotensin receptor antagonists may go beyond the blood pressure lowering effects of these agents (Bell 2003, Bui et al. 2003, Lewis & Lewis 2003). In this context it is noteworthy that angiotensin II can induce direct, pro-oxidative effects on the vascular endothelium mediated, at least in part, by intraendothelial reactive species formation via a new family of NAD(P)H oxidase subunits, known as the non-phagocytic NAD(P)H oxidase proteins. Reactive oxidant species produced following angiotensin II-mediated stimulation of NAD(P)H oxidases can exert direct oxidative effects, but can also signal through pathways such as mitogen-activated protein kinases, tyrosine kinases and transcription factors, and can lead to inflammation, hypertrophy, remodeling and angiogenesis (Cai et al. 2003). Recent work demonstrates that angiotensin II can also induce intraendothelial peroxynitrite formation (Wattanapitayakul et al. 2000, Mihm et al. 2003), as well as PARP activation (Szabó et al. 2004). Administration of angiotensin II triggers the activation of PARP in cultured endothelial cells in vitro, which is dose-dependently inhibited by PARP inhibitors, as well as by the compound apocynin, indicating the involvement of NAD(P)H oxidase-generated superoxide anions (Szabó et al. 2004). Angiotensin-induced PARP activation is also inhibited by L-NAME and diphe-nyleneiodonium (Szabó et al. 2004). Thus, angiotensin triggers the endothelial generation of reactive oxygen species from NAD(P)H oxidase, which react with constitutively produced NO, to produce peroxynitrite and other reactive nitrogen species, which induce DNA breakage and activate PARP in the vascular endothelium, leading to the development of endothelial dysfunction. This pathway also appears to be operative in vivo. Future work needs to establish the importance of this pathway in the context of diabetic complications. A human study analyzed forearm skin biopsy samples from healthy individuals with parental history of type 2 diabetes (T2DM), subjects with impaired glucose tolerance (IGT) and a group of type 2 diabetic patients and found that the percentage of PARP-positive endothelial nuclei was higher in the group of parental history of T2DM and diabetic patients when compared to the controls (Szabó et al. 2002b). In addition, significant correlations were observed between the percentage of PARP-positive endothelial nuclei and fasting blood glucose, resting skin blood flow, maximal skin vasodilatory response to the iontophoresis of acetylcholine (which indicates endothelium-dependent vasodilation) and nitrotyrosine immunostaining intensity. Nitrotyrosine immunoreactivity (a marker of reactive nitrogen species [e.g. peroxynitrite] formation) was also higher in the diabetic patients when compared to all other groups. Significant correlations were observed between nitrotyrosine immunostaining intensity and fasting blood glucose, HbA1c, intracellular adhesion molecule (ICAM), and vascular cellular adhesion molecule (VCAM). No differences in the expression of eNOS and RAGE were found among all four groups. The poly-morphism of the eNOS gene was also studied and was not found to influence eNOS expression or microvascular functional measurements. Thus, in humans, PARP activation is present in healthy subjects at risk of developing diabetes, as well as in established type 2 diabetic patients and it correlates with impairments in the vascular reactivity in the skin microcirculation (Szabó et al. 2002b). As interventional studies with PARP inhibitors in humans with diabetic endothelial dysfunction have not yet been conducted, it remains to be seen whether PARP activation in diabetic or prediabetic humans can be seen as a predictor or early marker for the development of diabetic vascular complications. PARP IN THE PATHOGENESIS OF DIABETIC CARDIOMYOPATHY, RETINOPATHY, NEPHROPATHY AND NEUROPATHY Cardiomyopathy - Superoxide-peroxynitrite-PARP pathway plays a key role in various models of myocardial ischemia-reperfusion injury (Zingarelli et al. 1998, Szabó et al. 2002). In diabetic rats the degree of myocardial infarction and the degree of myocardial contractile failure increases, but remains dependent on PARP activation (Xiao et al. 2004). The PARP pathway also plays pathogenetic role in the development of diabetic cardiomyopathy: cardiac dysfunction and PARP activation in the cardiac myocytes and the coronary vasculature develops both in streptozotocin-induced and genetic (nonobese diabetic) models of diabetes mellitus in rats and mice. Furthermore, treatment with the PARP inhibitor PJ34, starting one week after the onset of diabetes, restored normal vascular responsiveness and significantly improved cardiac function in diabetic mice and rats, despite the persistence of severe hyperglycemia. The beneficial effect of PARP inhibition persisted even after several weeks of the discontinuation of the PARP inhibitor treatment (Pacher et al. 2002d). The diabetic endothelial PARP pathway and the diabetic cardiomyopathy are possibly interrelated: the impairment of the endothelial function may lead to global or regional myocardial ischemia, which may secondarily impair cardiac performance. The protective effect of PARP inhibition against diabetic cardiac dysfunction extends several weeks beyond the discontinuation of treatment; this observation may have important implications for the design of future clinical trials with PARP inhibitors. The prolonged protective effect may be related to the permanent interruption by the PARP inhibitor of positive feedback cycles of cardiac injury. Indeed, previous studies in various pathophysiological conditions have demonstrated that PARP inhibitors suppress positive feedback cycles of adhesion receptor expression and mononuclear cell infiltration, as well as cellular oxidant generation (Zingarelli et al. 1998, Szabó et al. 2002, Ceriello et al. 2003a, Piconi et al. 2004). The mode of PARP inhibitors' cardioprotective action involves a conservation of myocardial energetics, as well as a prevention of the up-regulation of various pro-inflammatory pathways (cytokines, adhesion receptors, mononuclear cell infiltration) triggered by ischemia and reperfusion (Zingarelli et al. 1998, Szabó et al. 2002). It is conceivable that PARP inhibition exerts beneficial effects in experimental models of diabetic cardiomyopathy by affecting multiple pathways of injury, and by suppressing positive feedback cycles. Retinopathy - There is now evidence for PARP activation in the microvessels and ganglionic layer of the diabetic retina (Obrosova et al. 2004c). The causative role of PARP in diabetic retinopathy is now supported by two independent interventional preclinical studies. In one study (Zheng et al. 2004), a long-term (9-month) study was used to investigate the role of PARP in hyperglycemia-induced cell death in vitro, and in the deve-lopment of diabetic retinopathy in vivo. Streptozotocin-diabetic Lewis rats were treated with vehicle or the PARP inhibitor, PJ34. Diabetes was found to increase activity of PARP in retina measured at 2 months, and PJ34 inhibited this increase. PARP activation was detectable also in a subset of nuclei from retinal capillary endothelial cells and pericytes. Diabetes of 9 months duration significantly increased the number of both TUNEL-positive capillary cells and acellular capillaries (a marker of degenerate capillaries), and PJ34 significantly inhibited these alterations without influencing glycemic control. PJ34 also inhibited a diabetes-induced up-regulation of ICAM and leukostasis within the retinal vasculature. In a complimentary in vitro study, bovine retinal endothelial cells and pericytes were incubated in 5 mM (normal) and 25mM (elevated) glucose for 5 days with or without PJ34. High glucose significantly increased death of retinal capillary endothelial cells, and PARP inhibition prevented this cell death. In a second, independent study (Mabley et al. 2004), male C57/BL6 mice were rendered diabetic with a single injection of STZ. Diabetic mice, treated with the PARP inhibitor PJ34 for 6 months were investigated experimental retinopathy using retinal digest preparations and quantitative retinal morphometry. Diabetes over 6 months induced pericyte loss and increased the number of acellular capillaries. Treatment with PJ34 inhibited both, the loss of pericytes, and the formation of acellular capillaries. These data, taken together, suggest that hyperglycemia-induced PARP activation affects predominantly the retinal vasculature and is susceptible to pharmacological PARP inhibition. Nephropathy - The presence of glomerular depositions (mesangial distribution) of IgG was significantly reduced in streptozotocin-diabetic rats treated with the PARP inhibitor nicotinamide for 6 months (Wahlberg et al. 1985). In agreement with these results we have recently demonstrated that PARP activation is present in the tubuli of streptozotocin-induced diabetic rats. This PARP activation is attenuated by two structurally unrelated PARP inhibitors, 3-aminobenzamide (ABA) and 1,5-isoquinolinediol (ISO), which also counteracted the overexpression of endothelin-1 (ET-1) and ET receptors in the renal cortex (Minchenko et al. 2003). Neuropathy _ Recent studies suggest that the oxidative/nitrosative stress-PARP pathway also plays a key role in the development of diabetic neuropathy: the progressive slowing of sensory and motor neuron conductance in diabetic rats and mice is preventable by PARP inhibition or PARP deficiency, and this is associated with maintained neuronal phosphocreatine levels, as well as improved endoneurial blood flow (Cheng & Zochodne 2003, Obrosova et al. 2003, 2004a, Li et al. 2004). Importantly, pharmacological PARP inhibition is not only a preventive option: it can also restore sensory and motor neuronal conduction in already established diabetic neuropathy, at least in murine models of the disease (Li et al. 2004). In a recent study it was also demonstrated that PARP overactivation plays a role in diabetic neuropathy associated brain dysfunction (Kuchmerovska et al. 2004). It is important to re-emphasize that, at least in part, diabetic retinopathy, neuropathy and nephropathy all develop on the basis of endothelial dysfunction (Nitenberg 2002, Obrosova 2003) and it is conceivable that improvements in endothelial function may underlie some of the improvements seen in neuronal conduction in diabetes in response to PARP inhibition. CONCLUDING REMARKS Thus, we conclude that the PARP pathway plays important regulatory roles in the pathogenesis of vascular endothelial dysfunction in pathophysiological conditions associated with oxidative stress, including diabetes. It appears that some clinical or experimental therapeutic interventions, which are known to have some vascular protective effects in diabetes (antioxidant therapies, PPAR agonists, etc.) are able to suppress the activation of PARP in the cardiovascular system (Cuzzocrea et al. 2004, Da Ros et al. 2004), although whether such effects are also present in diabetic models remains to be seen. It is noteworthy in this respect that in preclinical studies, administration of the aldose reductase inhibitors sorbinil or fidarestat to diabetic rats not only corrected diabetes-induced depletion of glutathione and ascorbate, downregulation of SOD activity and accumulation of lipid peroxidation products in the peripheral nerve, counteracted superoxide formation in vasa nervorum and was effective against multiple indices of diabetes-associated retinal oxidative and nitrosative stress, but also inhibited poly(ADP-ribose) accumulation (a marker of PARP activation) in diabetic nerve and retina (Obrosova et al. 2004d). Similar results were obtained with FP15, a novel peroxynitrite decomposition catalyst compound (Szabó et al. 2002a). In a murine study, sciatic motor nerve conduction velocity and hind-limb digital sensory conduction velocity were reduced in diabetic mice versus controls, and both indices were normalized by FP15, which also ameliorated the accumulation of poly(ADP-ribose) accumulation in diabetic nerves (Obrosova et al. 2004b). The role of oxidative/nitrosative stress-PARP pathway (Figure) is not limited to the diabetes-induced vascular dysfunction but is also relevant for the pathogenesis of other diabetic complications, such as cardiomyopathy, retinopathy, nephropathy and neuropathy. PARP activation, thus, is a unique checkpoint in the development and progression of various diabetic complications. PARP inhibition may emerge as a novel approach for the prevention or reversal of diabetic complications. The benefits and potential risks associated with chronic administration of PARP inhibitors are discussed in a recent review (Southan & Szabó 2003). The future therapeutic utility of PARP inhibition for the experimental therapy of diabetic complications should be further explored by preclinical and clinical investigations. CONCLUSION We have reviewed here the current knowledge on ANXA1 biology with respect to cells involved in the inflammatory process. Despite brief descriptions of the pattern of expression of ANXA1 and its possible modulation by GC or inflammation, we have tried to place more emphasis on the pharmacological effects that have been ascribed to the protein and/or its peptido-mimetics. Withfew exceptions (e.g. the neutrophil and perhaps the epithelial cell) it is clear that much more must be done. The convenience of the bioactive ANXA1 N-terminal-derived peptides or the antiflammins could contribute towards defining specific pharmacological effects, that perhaps in the past have been hampered by the lack of recombinant protein. In addition, the recent notion that ANXA1 effects might be mediated by members of the FPR family, possibly ALXR (at least in human systems), may further aid the development of novel therapeutics based on the ANXA 1 pathway. REFERENCES
The following images related to this document are available:Photo images[oc05024f1.jpg] |
| |||||||||

{kind=link}