 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol.100, Suppl. 1, March, 2005, pp. 167-172 Macrophage elastase (MMP-12): a pro-inflammatory mediator? Soazig Nénan/*, Elisabeth Boichot*, Vincent Lagente*/+, Claude P Bertrand/++ Pfizer Global R&D,
Fresnes Laboratories, Fresnes, France *Laboratoire de Pharmacodynamie et
de Pharmacologie Moléculaire, INSERM U620, University of Rennes 1,
Rennes, 2, avenue du Professeur Léon Bernard, 35043
Rennes Cedex, France Received 8 November
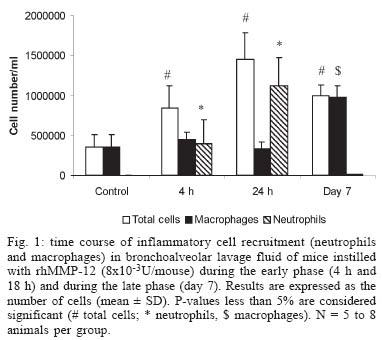
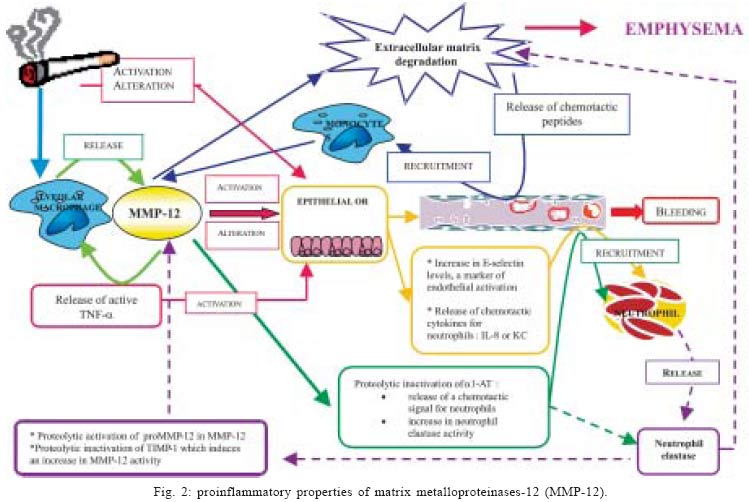
2004 Code number: oc05045 As many metalloproteinases (MMPs), macrophage elastase (MMP-12) is able to degrade extracellular matrix components such as elastin and is involved in tissue remodeling processes. Studies using animal models of acute and chronic pulmonary inflammatory diseases, such as pulmonary fibrosis and chronic obstrutive pulmonary disease (COPD), have given evidences that MMP-12 is an important mediator of the pathogenesis of these diseases. However, as very few data regarding the direct involvement of MMP-12 in inflammatory process in the airways were available, we have instilled a recombinant form of human MMP-12 (rhMMP-12) in mouse airways. Hence, we have demonstrated that this instillation induced a severe inflammatory cell recruitment characterized by an early accumulation of neutrophils correlated with an increase in proinflammatory cytokines and in gelatinases and then by a relatively stable recruitment of macrophages in the lungs over a period of ten days. Another recent study suggests that resident alveolar macrophages and recruited neutrophils are not involved in the delayed macrophage recruitment. However, epithelial cells could be one of the main targets of rhMMP-12 in our model. We have also reported that a corticoid, dexamethasone, phosphodiesterase 4 inhibitor, rolipram and a non-selective MMP inhibitor, marimastat could reverse some of these inflammatory events. These data indicate that our rhMMP-12 model could mimic some of the inflammatory features observed in COPD patients and could be used for the pharmacological evaluation of new anti-inflammatory treatment. In this review, data demonstrating the involvement of MMP-12 in the pathogenesis of pulmonary fibrosis and COPD as well as our data showing a pro-inflammatory role for MMP-12 in mouse airways will be summarized. Key words: matrix metalloproteianse - metalloelastase - airway inflammation - chronic obstructive pulmonary disease Chronic obstructive pulmonary disease (COPD) is one of the major causes of mortality and morbidity across the world and its prevalence is still increasing (Pauwels 2000, Petty 2000). The major triggering factor is cigarette smoking, which accounts for 80-90% of the COPD cases. However, in the population of smokers, only 15% of the subjects develop chronic airflow limitation (Saetta 1999). COPD is characterized by the presence of a partially reversible airflow obstruction. This pathology is also associated with an airway inflammatory process characterized by an accumulation of inflammatory cells such as macrophages and neutrophils. Indeed, it has been shown that cigarette smoke consistently produces an increase in the neutrophil number in bronchoalveaolar lavage fluid and in tissue (Ludwig et al. 1985, Eidelman et al. 1990, Finkelstein et al. 1995). Macrophage numbers are also elevated in the lungs of smokers and patients with COPD where they accumulate in the alveoli, bronchioli and small airways. Furthermore, there is a positive correlation between macrophage number in the alveolar walls and the mild-to-moderate emphysema status in patients with COPD (Tetley 2002). It is generally believed that the development of emphysema reflects a relative excess of cell-derived proteases that degrade the connective tissue of the lung and a relative paucity of antiproteolytic defenses. This theory is often referred to as the "protease-antiprotease imbalance" hypothesis and involves mainly serine proteases like neutrophil elastase and matrix metalloproteinases (MMPs). MMPs is a family of structurally related extracellular matrix (ECM)-degrading enzymes that are collectively capable of degrading essentially all ECM components and that can further be subdivided in collagenases (MMP-1, -8, -13), gelatinases (MMP-2, -9), stromelysins (MMP-3, -10, -11), matrilysin (MMP-7), macrophage metalloelastase (MMP-12),membrane-type MMPs (MMP-14, -15, -16, -17) and other MMPs (Shapiro 1998). Among MMPs, MMP-12 is a 54 kDa proenzyme that is processed into a 45 kDa and then a 22 kDa active forms. The human gene, which is designated human macrophage metalloelastase, produces a 1.8-kb transcript encoding a 470-amino acid protein that is 64% identical to the mouse protein. Both the mRNA and protein were detected in alveolar macrophages. As in the mouse, the predicted human 54-kD protein is processed by loss of both N- and C-terminal residues to a 22-kD mature form (Shapiro et al. 1993). MMP-12 is mainly produced by macrophages and has been shown to be associated with inflammatory skin diseases (Saarialho-Kere et al. 1999, Vaalamo et al. 1999, Suomela et al. 2001), atherosclerosis (Matsumoto et al. 1998), aneurysms (Curci et al. 1998) and cancers (Cornelius et al. 1998, Kerkela et al. 2000, 2001). MMP-12 seems also to be clearly involved in acute and chronic pulmonary inflammatory diseases associated with an intense airway remodeling such as COPD. Indeed, it has been suggested that MMP-12 gene polymorphism may account for this disease variability and is one of the causative factors in smoking related injury (Belvisi et al. 2003). The aim of this review is to summarize data exploring the role of MMP-12 in inflammatory pulmonary diseases and to delineate its involvement in the tissue remodeling process and in the inflammatory events observed in COPD. MMP-12 AND INFLAMMATORY PULMONARY DISEASES The involvement of MMP-12 in inflammatory pulmonary diseases has been mainly studied using animal models. It appears that this metalloelastase could be an important mediator in the pathogenesis of acute lung injury and chronic lung injury. Pulmonary fibrosis corresponds to the end stage of acute lung injury. The administration of bleomycin in rodent airways causes similar inflammatory and fibrotic responses observed in patients with pulmonary fibrosis. During the acute phase of this bleomycin-induced injury, a significant activation of MMP-12 has been observed in mice and rats (Koslowski et al. 1998, Swiderski et al. 1998). It has also been shown that proMMP-12 is converted in its active form early after bleomycin treatment during the peak time of macrophage levels and is associated only with areas of hemorrhage (Swaisgood et al. 2000). In an immune complex-induced acute lung injury model using mice containing a target disruption of the MMP-12 gene (MMP-12 KO mice), neutrophil influx into the alveolar space and lung permeability in KO mice has been reduced by 50% of that observed in wild-type littermates. These results has been correlated with histological evidence of reduced injury in the MMP-12 KO mice (Warner et al. 2001). In addition, MMP-12 seems to play a predominant role in the pathogenesis of chronic lung injury and particularly in emphysema. Indeed, MMP-12 is able to degrade different substrates among which elastin (Gronski et al. 1997). Elastin represents about 2.5% (wt/wt) of the dry weight of the lung and is distributed widely throughout the lungs (Starcher 1986). This protein is vital for the elastic recoil of the small airways and their ability to resist negative pressure collapse. In emphysema, elastin content of the lung parenchyma is decreased (Wright 1995) and ultrastructurally, elastic fibers are disorganized and probably nonfunctional (Shapiro 2000). Moreover, elastin degradation products, such as desmosine, are increased in the urine of subjects with COPD (Stone et al. 1995) and correlate with the rate of lung function decline (Gottlieb et al. 1996). In vitro studies on alveolar macrophages collected from COPD patients have shown their ability to degrade more elastin than macrophages collected from healthy volunteers (Russell et al. 2002). Using immunocytochemistry, we have previously observed MMP-12-positive macrophages in both COPD and control samples. However, the number of MMP-12 expressing-macrophages together with the staining intensity was higher in bronchoalveolar lavage (BAL) samples from COPD patients than in controls subjects. Similar results were noted in bronchial biopsies with higher MMP-12 expression in COPD subjects than in controls. Enhanced MMP-12 activity was also shown in BAL fluids from patient with COPD in comparison with control subjects. This study demonstrated that COPD patients produce greater quantities of MMP-12 than controls, which may be a critical step in the pathogenesis of COPD and emphysema (Molet et al. 2004). Studies using MMP-12 KO mice have demonstrated that macrophage recruitment in lungs and emphysema induced by long-term exposure to cigarette smoke were linked to MMP-12 (Hautamaki et al. 1997). MMP-12 KO mice were subjected to cigarette smoke over a 6 months period. In contrast to wild-type mice, MMP-12 KO mice did not have increased numbers of macrophages in their lungs and did not develop emphysema. The monthly intratracheal instillation of monocyte chemoattractant protein (MCP)-1 in the lungs of MMP-12 smoke-exposed KO mice caused an increase in macrophage recruitment. However, despite the presence of the macrophages, these MMP-12 KO mice did not develop air space enlargement in response to smoke exposure. These data suggest that MMP-12 is probably sufficient for the development of emphysema that results from chronic inhalation of cigarette smoke (Hautamaki et al. 1997). The macrophage recruitment observed in response to cigarette smoke could be linked to the elastolytic properties of MMP-12. Indeed, MMP-12 generates elastin-derived peptides and experiments realized in modified Boyden chambers have shown that these elastin-derived peptides are chemotactic for monocytes (Senior et al. 1980). In a more recent study, it was reported that inflammatory lesions in the lungs of mice contained significantly more MMP-12 in macrophages at 10, 20, and 30 days of cigarette smoke exposure than in controls of mice exposed to 60 days (Valença et al. 2004). These results suggest that elastin degradation took place during development of pulmonary change in mice exposed to cigarette smoke and activation of MMPs specific to elastin may be a determining factor for susceptibility to emphysema (Valença et al. 2004). Through a global analysis of pulmonary gene expression in the lungs of mice lacking integrin beta-6, using oligonucleotide arrays, Kaminski et al. (2000), have identified a marked induction of MMP-12. More recently, Morris et al. (2003) have demonstrated that Itgb6-null mice develop age-related emphysema that is completely abrogated either by transgenic expression of the beta-6 integrin sub-unit that supports TGF-β activation, or by loss of MMP-12. Furthermore, this study has showed that the effects of Itgb6 deletion are overcome by simultaneous transgenic expression of active TGF-β1. This suggests that the loss of integrin-mediated activation of latent TGF-β causes age-dependent pulmonary emphysema through alterations of macrophage MMP-12 expression. Finally, a functional alteration in the TGF-β activation pathway affects the susceptibility to this disease. In an acute model of smoke exposure, neutrophils, desmosine and hydroxyproline, markers for elastin and collagen breakdown respectively, were examined in BAL fluids of MMP-12 KO mice and wild-type mice at 24 h after smoke exposure. None of these markers could be detected in MMP-12 KO mice, suggesting that acute smoke-induced connective tissue breakdown, the initial step to emphysema, requires both neutrophils and MMP-12 and that the neutrophil influx is dependant on the presence of MMP-12 (Churg et al. 2002). In the same model, compared to wild-type littermates, MMP-12 KO mice showed impaired TNF-α release after acute smoke exposure. Levels of E-selectine, a specific marker of endothelial activation, were increased in wild-type mice but not in MMP-12 KO mice after smoke exposure. Taken together, these data indicate that MMP-12 could mediate smoke-induced inflammation by releasing TNF-α from macrophages, with subsequent endothelial activation, neutrophil influx and proteolytic matrix breakdown (Churg et al. 2003). The cross-talk between neutrophils and macrophages and the relative involvement of neutrophil elastase and MMP-12 were clarified in a long-term smoke exposure model. After 6 months of smoke exposure, mice that were deficient for neutrophil elastase (NE KO mice) were significantly protected from the development of emphysema and MMP-12 KO mice were totally protected, suggesting a significant role of these proteases in the development of emphysema in mice. Moreover interactions between the neutrophil elastase and the MMP-12 proteolytic systems were observed, with each augmenting the other's destructive capacity. Indeed, MMP-12 may degrade the serine protease inhibitor a1-antitrypsine and neutrophil elastase may degrade the tissue inhibitor of MMP, (TIMP)-1. Neutrophil elastase may also be required for the proteolytic activation of pro-MMP-12. The absence of neutrophil elastase had dramatic effects on both neutrophil and macrophage accumulation in the lungs in response to cigarette smoke. MMP-12 KO mice had also reduced monocyte recruitment following smoke exposure. Hence, the protection from emphysema in smoke-exposed NE KO mice could be linked to a decrease in the level of active MMP-12 because of the requirement of neutrophil elastase for macrophage accumulation in the lungs and because of the activation of proMMP-12 by this serine protease (Shapiro et al. 2003). It has been hypothesized that some of the mediators involved in the inflammatory process observed in emphysematous tissues could directly induce emphysema. Interferon (IFN)-γ has been one of those candidates. Indeed, CD8+ lymphocytes infiltration is a prominent feature of inflammation observed in COPD patients and these cells are known to produce IFN-γ. In transgenic mice that overexpress IFN-γ, a phenotype that mimics human emphysema can be observed. In this model, the IFN-γ overexpression induced an increase in macrophage, lymphocyte and neutrophil numbers in BAL fluids and in lungs. Moreover, IFN-γ shifted the pulmonary protease/ antiprotease balance in a proteolytic direction via the induction of MMP-12 and a variety of cathepsins. These observations suggest that cigarette smoke may induce MMP-12 via the induction of IFN-γ(Wang et al. 2000). Transgenic mice that overexpress the interleukin (IL)-13 mimic some of the features observed in COPD patients. They develop a neutrophil, eosinophil, and macrophage cell-rich lung inflammation associated with MMPs and cathepsins induction, alveolar enlargement and enhanced pulmonary compliance (Zhu et al. 1999, Zheng et al. 2000). It has been demonstrated that IL-13 overexpression induces all these events via a MMP-9 and MMP-12 dependant mechanism. The induction of MMPs-2, -9,-13 , -14 induced by IL-13 is mediated, in part, by a MMP-12 dependant pathway and MMP-12 makes a crucial contribution to the accumulation of eosinophils and macrophages (Lanone et al. 2002). Hence, taken together, these data show clearly that MMP-12 play a pivotal role in the tissue remodeling process in pulmonary inflammatory diseases. Despite evidences concerning its ability to generate a chemotactic signal for inflammatory cells such as monocytes/macrophages, very few data regarding the inflammatory potential of MMP-12 are available. In their study, Churg et al. (2003) have suggested that macrophages could be one of the target for MMP-12, that could release TNF-α from macrophages and initiate a cascade of inflammatory process. The direct involvement of MMP-12 in the development of the inflammatory process in the airways has also been explored in recent studies. PROINFLAMMATORY PROPERTIES OF MMP-12 The direct effect of MMP-12 in the development of inflammatory process in mouse airways has been evaluated using a recombinant form of human MMP-12 (rhMMP-12). A single instillation of rhMMP-12 in mouse airways elicited an intense inflammatory response characterized by the development of two successive phases. Indeed, rhMMP-12 induced an acute, severe and specific recruitment of neutrophils reaching a peak at 18 h. The number of macrophages remained stable throughout this period (Fig. 1) (Nénan et al. 2004a). The recruitment of neutrophils was associated with a very transient increase in cytokine and chemokine levels (TNF-α, MIP-1α, MCP-1, IL-6, and KC) in BAL fluids and in lung homogenate supernatants. An increase in gelatinase (MMP-2 and MMP-9) activities in BAL fluid during the early phase was also observed. This increase in inflammatory mediators was transient during the first 24 h with a maximum of activity between 4 and 8h. Then, a delayed phase, from day 4 to day 15 post instillation, was observed with a significant and specific macrophage influx, stable over a period of 10 days without any other studied inflammatory signals. The role played respectively by resident alveolar macrophages, by recruited neutrophils and by epithelial cells in the delayed recruitment of macrophages induced by rhMMP-12 was investigated (Nénan et al. 2004b). Mice depleted of circulating neutrophils, using a cytotoxic antibody, did not present any increase in neutrophil numbers in BAL fluids, 4 and 24 h after rhMMP-12 instillation. However, the macrophage recruitment was not modified as compared to control mice at day 7. Similar results were obtained when the gene for neutrophil elastase was knocked out in mice. Intranasal instillation of clodronate liposomes, 72 h prior to rhMMP-12 instillation, induced macrophage depletion. This treatment did not modify the macrophage recruitment at day 7. Moreover, the stimulation of mouse macrophages by rhMMP-12 in vitro did not elicit the release of cytokines in culture supernatants. In contrast, the in vitro stimulation of A549 epithelial cells induced the release of IL-8 (Nénan et al. 2004b). These results suggest that resident alveolar macrophages and recruited neutrophils do not play a role in the delayed macrophage recruitment induced by rhMMP-12. However, epithelial cells could be the initial target for rhMMP-12 leading to the inflammatory process observed in our model. PHARMACOLOGICAL MODULATION OF rhMMP-12-INDUCED INFLAMMATION As lung inflammation induced by rhMMP-12 instillation in mouse airways partially mimics some of the COPD features, drugs with a potential efficacy in COPD were tested in this model (Nénan et al. 2004a). Hence the profile of activity of two classes of anti-inflammatory agent, the corticosteroid, dexamethasone, and the selective PDE4 inhibitor, rolipram, was determined. A non-specific MMP inhibitor, marimastat, was also tested on neutrophil influx associated with cytokine release and increase in MMP-9 activity and on the delayed macrophage recruitment. Marimastat (100 mg/kg), dexamethasone (10 mg/kg), and rolipram (0.1 and 0.3 mg/kg), administered orally 1 h before rhMMP-12 instillation, were able to significantly decrease neutrophil recruitment at 4 and 24 h. Only marimastat (30 and 100 mg/kg) was effective on the macrophage recruitment at day 7. In BAL fluids, marimastat (100 mg/kg), dexamethasone (10 mg/kg) and rolipram (0.3 mg/kg) significantly decreased IL-6, KC (IL-8), macrophage inflammatory protein (MIP)-1a and MMP-9 levels. Similar results were observed in lung homogenates except for rolipram, which was ineffective. Hence, a corticosteroid, a PDE4 inhibitor and a non-selective MMP inhibitor were able to reverse some of these inflammatory events. Taken together, these data indicate that this MMP-12-induced inflammatory model could highlight some of the inflammatory response seen in COPD and could be used for the pharmacological evaluation of new anti-inflammatory mechanism of action. CONCLUDING REMARKS The underlying mechanisms of emphysema include inflammatory and remodeling processes in the airways. Because of its ability to induce an inflammatory response and tissue remodeling, it may be possible to consider MMP-12 as an essential component of the process leading to the development of the disease (Fig. 2). Hence, MMP-12 could be pivotal in the two main hypotheses that are proposed to explain the pathological process of COPD i.e. the "elastase/antielastase imbalance" theory and the "inflammation/repair" theory. Moreover, as several drugs with a potential efficacy in COPD were able to reverse some of the inflammatory events induced by MMP-12, these observations emphasize that MMP-12 could be considered as a potential target in the COPD treatment.
Copyright 2005 Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc05045f2.jpg] [oc05045f1.jpg] |
| |||||||||

{kind=link}
{kind=link}