 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol.100, Suppl. 1, March, 2005, pp. 173-176 Protease-activated receptors and inflammatory hyperalgesia Nathalie Vergnolle Department of Pharmacology
and Therapeutics, Faculty of Medicine, University of Calgary, 3330 Hospital
Drive NW Calgary, T2N4N1, Canada Received 8 November
2004 Code number: oc05046 Recent advances in basic science pointed to a role for proteinases, through the activation of proteinase-activated receptors (PARs) in nociceptive mechanisms. Activation of PAR1, PAR2 and PAR4 either by proteinases or by selective agonists causes inflammation inducing most of the cardinal signs of inflammation: swelling, redness, and pain. Sub-inflammatory doses of PAR2 agonist still induced hyperalgesia and allodynia while PAR2 has been shown to be implicated in the generation of hyperalgesia in different inflammatory models. In contrast, sub-inflammatory doses of PAR1 increases nociceptive threshold, inhibiting inflammatory hyperalgesia, thereby acting as an analgesic agent. PARs are present and functional on sensory neurons, where they participate either directly or indirectly to the transmission and/or inhibition of nociceptive messages. Taken together, the results discussed in this review highlight proteinases as signaling molecules to sensory nerves. We need to consider proteinases and the receptors that are activated by proteinases as important potential targets for the development of analgesic drugs in the treatment of inflammatory pain. Key words: proteases - inflammation - pain - thrombin - trypsin - tryptase Proteases can signal to cells through a variety of mechanisms. They can transform a pro-receptor or a pro-agonist into an active receptor or agonist respectively, by cleaving these molecules. They can act as any other agonist by binding to a receptor through their non-catalytic sites. Compelling evidence that has accumulated in recent years also indicates that certain proteases, such as thrombin, tryptase, and trypsin, can signal to cells through the activation of protease-activated receptors (PARs). PARs are activated by a unique mechanism that involves the proteolytic cleavage of their N-terminal extracellular domain. This cleavage, due to the action of diverse proteases, releases a new N-terminal domain that acts as a tethered ligand, binding the receptor itself on its second extracellular loop, to induce an intracellular signal (Fig. 1). Four members of the PARs family have been cloned. PAR1, PAR3 and PAR4 are considered as thrombin receptors since they have been shown to be responsible for thrombin-induced platelet activation. However, these receptors can also be activated by other proteases such as trypsin and cathepsin G for PAR4, coagulation factors Xa and VIIa for PAR1 (Fig. 1). Another member of this family, PAR2, is not activated by thrombin, but can be activated by trypsin and mast cell tryptase. Useful pharmacological tools have been raised to specifically activate those receptors: small synthetic peptides corresponding to the tethered ligand domain are able to activate selectively PAR1, PAR2 and PAR4. Surprinsingly, PAR3 cannot be activated by peptidic sequences corresponding to its tethered ligand domain, rendering the study of the physiological role of this receptor more difficult in the absence of selective agonist. The peptidic sequence corresponding to the human PAR2 receptor, SLIGKV-NH2 (where each letter correspond to the amino acid code), selectively activates the receptor (Fig. 1), as does the SLIGRL-NH2 peptide corresponding to the rat sequence. The tethered ligand sequence corresponding to the human PAR1 receptor (SFLLR-NH2; Fig. 1) is not selective for PAR1, but can also activate PAR2. A substitution of the serine amino acid by a threonine (TFLLR-NH2) remarkably increases the specificity of the peptide for the PAR1 receptor, and this later peptide is now used as a selective PAR1 agonist. Peptide corresponding to the tethered ligand of PAR4 (GYPGKV-NH2; Fig. 1) is specific for PAR4, but is not a very potent agonist. Substitution of the first amino acid by an Alanine residue (AYPGKV-NH2) considerably increases the potency of the agonist for PAR4 activation and is now preferably used as a PAR4 agonist. PARs AND INFLAMMATION Several studies have shown the pro-inflammatory effects of acute activation of PAR2. First, we have shown that intraplantar injection of the selective PAR2-activating peptide SLIGRL-NH2 caused edema and inflammatory cell recruitment (Vergnolle et al. 1999a). Later, we have demonstrated that PAR2 activation promotes the first signals for leukocyte recruitment to the site of inflammation, causing leukocyte rolling, adhesion and translocation across the wall of blood vessels (Vergnolle 1999). In the skin, acute activation of PAR2 leads to skin inflammation and PAR2 activation has been implicated in the generation of inflammatory signs associated with contact dermatitis (Seeliger et al. 2003). A recent study by Ferrell et al. (2003), has also demonstrated a prominent role for PAR2 in an animal model of monoarthritis, PAR2-deficient mice did not developed signs of chronic inflammation. In the gut, acute activation of PAR2 caused colitis characterized by gut wall edema, granulocyte recruitment, increased permeability and release of pro-inflammatory cytokines such as interleukin-1 and TNF-α (Cenac et al. 2002). In the airways, the role of PAR2 is controversial. Although studies have shown that PAR2-activating peptides caused relaxation of isolated airways and was protective against bronchoconstrictor challenge (Cocks et al. 1999), other studies have shown that mice that lack functional PAR2 showed extensive allergic response compared to wild-type mice, suggesting a prominent pro-inflammatory role for PAR2 activation in airway diseases (Schmidlin et al. 2002). Similar controversies also exist in the gut, where chronic and systemic treatment with PAR2 agonist was protective against a chronic model of inflammatory bowel disease (Fiorucci et al. 2001), while acute activation of PAR2 in colonic tissues led to inflammation (Cenac et al. 2002). Overall, PAR2 is present in many cells involved in inflammation (endothelial cells, mast cells, neutrophils, eosinophils, epithelium, etc.), and its activation on those cells provokes the release of many inflammatory mediators (prostaglandins, nitric oxide, cytokines, etc.), further supporting the idea that PAR2 activation plays a prominent role in inflammatory pathologies (Vergnolle 2000, 2001b). Several studies have also shown a pro-inflammatory role for PAR1 activation (Cirino et al. 1996,Vergnolle et al. 1999b), and most recently, we have demonstrated that PAR4 agonists induced oedema and granulocyte infiltration when injected into the rat paw (Hollenberg et al. 2004). PARs ON SENSORY NEURONS Investigating the mechanisms of PAR2-induced inflammation, we have discovered that the receptor was present on sensory nerves, where its activation was able to cause the release of neuropeptides such as substance P and CGRP (Steinhoff et al. 2000). This PAR2-induced neuropeptide release was responsible for the edema observed after intraplantar injection of PAR2 agonists, but not for granulocyte recruitment to the site of inflammation (Steinhoff et al. 2000). Other studies have shown that the effects of PAR2 agonists on chloride secretion, mucus secretion in the gastro-intestinal tract, or coronary vasodilatation, were mediated by a mechanism involving activation of C-fibers (Green et al. 1999, Kawabata et al. 2001, McLean et al. 2002). These results suggest that direct activation of PAR2 on sensory neurons, and particularly on C-fibers, is responsible for different pathophysiological changes associated with inflammation, including a neurogenic inflammatory response. PAR1 was also found present on sensory neurons (de Garavilla et al. 2001), but its activation failed to release neuropeptides in the different tissues observed. However, neuropeptide receptor antagonists were able to significantly reduce PAR1 agonist-induced oedema (de Garavilla et al. 2001). Taken together, these results suggest that PAR1 activation leads to inflammation through a pathway that involves a neurogenic mechanism that might be indirectly activated after PAR1 activation. The role of PAR1 activation in inflammation seems to be linked to activation of PAR1 on other cell types than sensory neurons. PAR4 was also found present on peripheral neurons (D'Andrea et al. 2003). However, the functionality of PAR4 on those cells has never been investigated. INVOLVEMENT OF PAR2 ACTIVATION IN INFLAMMATORY HYPERALGESIA The presence and functionality of PAR2 on sensory neurons led us to investigate whether or not PAR2 activation was implicated in inflammatory nociceptive pathways. Since PAR2 agonists caused inflammation, we first defined doses of PAR2 agonists that did not cause any signs of inflammation, by following oedema, granulocyte recruitment, prostaglandin release and blood flow (Vergnolle et al. 2001a). Then, we used this sub-inflammatory dose to investigate whether or not it was capable of inducing hyperalgesia in response to a thermal or mechanical stimulation. Intraplantar injection of PAR2-activating peptide, trypsin or mast cell tryptase at doses that did not cause inflammation, provoked thermal and mechanical hyperalgesia. The same injections also caused activation of nociceptors at the spinal level, as followed by an increased fos expression in the superficial laminae (I and II) of the dorsal horn (Vergnolle et al. 2001a). Moreover, this study showed that PAR2-deficient mice developed significantly less inflammatory hyperalgesia in response to intraplantar injection of formalin or the mast cell degranulator compound 48/80 (Vergnolle et al. 2001a). The PAR2 agonist-mediated hyperalgesia was dependent on a mechanism involving central activation of neurokinin-1 receptors, release of pre-pro tachykinins and release of prostaglandins (Vergnolle et al. 2001a). More recently, we have shown that activation of PAR2 was able to potentiate responses of TRPV1 receptors to capsaicin (Amadesi et al. 2004). In that study, we showed through both a pharmacological and gene-deletion approach that TRPV1 was implicated in PAR2-induced thermal hyperalgesia. We showed that PAR2-induced thermal hyperalgesia was inhibited by the TRPV1 antagonist capsazepine, and completely abolished in TRPV1-deficient mice, while PAR2-induced mechanical hyperalgesia was not changed by TRPV1 deficiency or antagonist treatments. Further, this study showed that TRPV1 activation in dorsal root ganglia neurons was potentiated by pre-exposure of those neurons to PAR2 agonists, and this potentiating mechanism was PKC-dependent (Amadesi et al. 2004). Other studies also suggest the involvement of PAR2 in visceral pain (Hoogerwerf et al. 2001, Coelho et al. 2002). Colonic or pancreatic activation of PAR2 was shown to be responsible for activation of nociceptors at a spinal level and in the case of colon, PAR2 agonists were able to induce long-lasting visceral hyperalgesia. In vitro studies showing that activation of PAR2 on peripheral neurons provoked calcium mobilization (Steinhoff et al. 2000), but also long-lasting hyperexcitability (Reed et al. 2003), suggest that the hyperalgesic effects of PAR2 agonists are due to the direct activation of the receptor on sensory neurons. However, the fact that PAR2 agonists provoke the release of different mediators (prostaglandins, cytokines, etc.) in different cell types (endothelial cells, leukocytes, etc.) needs also to be considered in the context of inflammatory pain. PAR2 activation might also participate to inflammatory pain by inflammatory mediators-induced sensitization of sensory neurons. THROMBIN RECEPTOR ACTIVATION: NEW ANALGESIC PATHWAYS? Peripheral activation
(intraplantar injections) by thrombin or a selective activating peptide of
the thrombin receptor PAR1, provoked an increase in nociceptive
threshold to thermal and mechanical stimulus (Asfaha et al. 2002). This anti-nociceptive
effect observed in basal conditions was reproduced in inflammatory conditions
after carrageenan intraplantar injection (Asfaha et al. 2002). Another study
has shown recently that spinal activation of PAR1 inhibited NMDA-induced
nociceptive activity through a mechanism dependent on endothelin A (Fang
et al. 2003). Because PAR1 activation on sensory neurons causes
calcium mobilization, one can doubt that PAR1 agonists exert their
anti-nociceptive effects by direct activation of sensory fibers. Further
studies on the signaling pathways and electrical responses of sensory neurons CONCLUSIONS Studies have shown
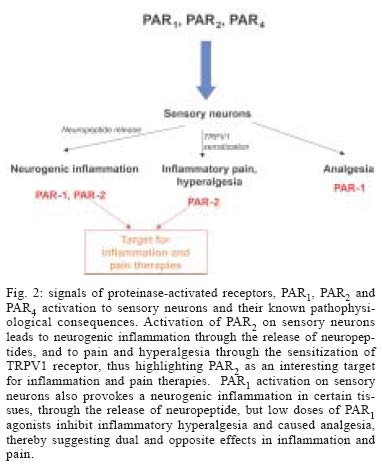
that PAR1, PAR2 and PAR4 are present on
peripheral neurons. The fact that PAR2 activation can induce neurogenic
inflammation and participate to the generation of inflammatory pain present
PAR2 as a valuable target for the treatment of inflammation and
pain (Fig. 2). PAR1 activation also
leads to inflammation with a neurogenic component. However, PAR1 activation
is also associated with analgesic response and inhibition of inflammatory
hyperalgesia, presenting PAR1 as a mediator that could exert dual
activity in inflammatory pain mechanisms. REFERENCES
Copyright 2005 Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc05046f2.jpg] [oc05046f1.jpg] |
| |||||||||

{kind=link}
{kind=link}