 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol.100, Suppl. 1, March, 2005, pp. 199-203 Epithelial cell signaling responses to enterohemorrhagic Escherichia coli infection Peter JM Ceponis++,
Jason D Riff+++, Philip M Sherman+ Received 8 November
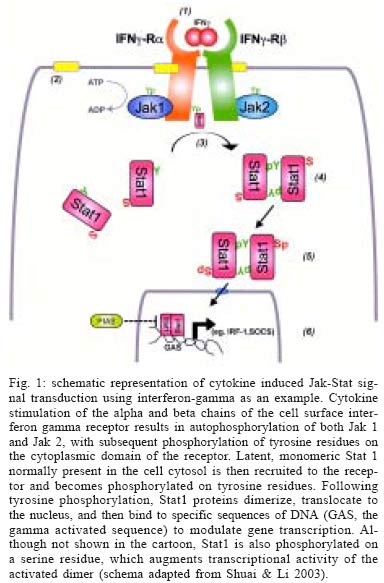
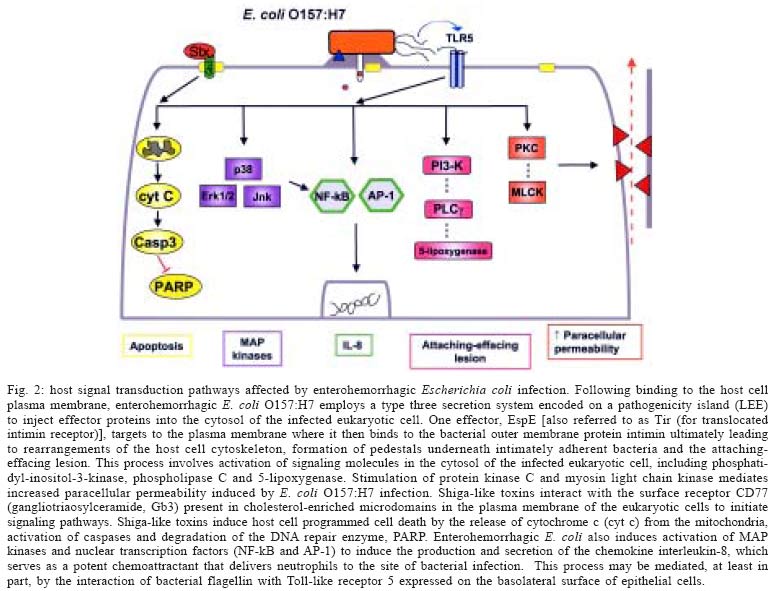
2004 Code number: oc05051 Enterohemorrhagic Escherichia coli, including the serotype O157:H7 that is most commonly identified with human disease, cause both sporadic cases and outbreaks of non-bloody diarrhea and hemorrhagic colitis. In about 10% of infected subjects, the hemolytic uremic syndrome (hemolytic anemic, thrombocytopenia, and acute renal failure) develops, likely as a consequence of systemic spread of bacterial-derived toxins variously referred to as Shiga-like toxin, Shiga toxin, and Verotoxin. Increasing evidence points to a complex interplay between bacterial products - for example, adhesins and toxins - and host signal transduction pathways in mediating responses to infection. Identification of critical signaling pathways could result in the development of novel strategies for intervention to both prevent and treat this microbial infection in humans. Key words: adherence - apoptosis - cytokine - O157:H7 - Shiga-like toxin - Verotoxin Microbial infections are characterized by a stereotypic pattern of host inflammatory responses to the infectious insult (Lawrence et al. 2002). In the intestinal tract, multiple invasive pathogens, including bacteria, parasites and viruses, may induce an enterocolitis in the infected host. Increasingly, evidence indicates that non-invasive gastrointestinal pathogens also are capable of inducing host inflammatory responses by pirating signaling transduction events in infected epithelial cells (Hecht 2001). Escherichia coli O157:H7 infection was first identified in the early 1980's in association with two outbreaks of hemorrhagic colitis in the United States of America. Infection was associated with the ingestion of under-cooked hamburger (Thorpe 2004). E. coli O157:H7 has been characterized as a non-invasive enteric pathogen that elaborates phage-encoded toxins variously referred to as Shiga-like toxins, Shiga toxins, and Vero cytotoxins. Subsequent studies showed that multiple E. coli serotypes produce Shiga-like toxins, and that many, but not all, of the various serotypes have been associated with human disease. Isolates associated with human disease are also referred to as enterohemorrhagic E. coli (EHEC) to distinguish them from other serotypes of Shiga-like toxin-producing E. coli that have not been demonstrated to cause disease in man. In addition to the cytotoxins, virulence determinants, such as attachment factors, are encoded in the bacterial genome of EHEC (Kaper et al. 2004). The environmental reservoir for EHEC is the anorectum of ruminants, including cattle (Naylor et al. 2003). These animals are generally asymptomatic. Moreover, one group of investigators recently provided provocative evidence indicating that carriage of Shiga-like toxin-producing E. coli benefits the ruminant, since toxin binding to cells infected with an oncogenic virus, but not to uninfected cells, leads to programmed cell death of the virus-infected cell (Menge et al. 2004). Fecal contamination of meats, fruits, and vegetables (e.g. non-pasteurized apple cider, radish sprouts), water supplies (including unchlorinated drinking water, swimming pools, and lakes) each have been reported as routes of transmission (Kassenborg et al. 2004). Person to person transmission of E. coli O157:H7 also has been described in both day care and hospital settings (Thorpe 2004). Children visiting petting zoos and farms are also at an increased risk of infection due to exposure to fecal contamination of the local environment (Crump et al. 2002). Karmali and colleagues (1983) were the first investigators to demonstrate that most cases of the hemolytic uremic syndrome characterized by a microangiopathic hemolytic anemia, low platelet count, and acute renal failure are due to EHEC infection. This is a major health concern since the hemolytic uremic syndrome is the most common cause of acute renal failure in children, and since current therapy is only supportive in nature. Evidence suggests that both anti-diarrheal agents and antimicrobial agents, in fact, may be harmful (Wong et al. 2000). Antibiotics might, for example, release a shower of pre-formed toxin from the periplasm of bacteria present in the gut lumen, thereby resulting in increased toxin delivery into the systemic circulation and presentation to susceptible vascular endothelial cells in the renal glomerulus. Accordingly, a better understanding of the pathobiology of disease is required in order to develop novel strategies to interrupt the infectious process. In animal models, including rabbits, gnotobiotic piglets, calves, greyhound dogs and antibiotic pre-treated mice, challenge with EHEC induces colonic inflammation with a predominance of neutrophils infiltrating into the lamina propria of the colon. Infusion of CD11/CD18 antibody reduces the host inflammatory response to E. coli O157:H7 infection of infant rabbits. Biopsies of colonic mucosa from infected humans also demonstrate a predominant neutrophilic infiltrate, with evidence of pseudomembrane formation comparable to that observed in Clostridium difficile-induced pseudomembranous colitis. This review will highlight selected aspects of host cell signal transduction responses to infection with EHEC that have been studied in our laboratory. The focus will be on apoptosis of epithelial cells in response to exposure to Shiga-like toxins and the inhibition of Stat (signal transducer and activation of transduction) signaling in epithelia infected with EHEC. The cited studies have employed epithelial cells (HEp-2 cells and T84) infected with bacteria using an in vitro reductionist model. BACTERIAL VIRULENCE FACTORS The entire genome of two E. coli O157:H7 strains, one originally isolated in the United States and the second in Japan, have been sequenced. These genomes have been compared to those of the laboratory E. coli K12 strain and a variety of other disease-related isolates, including enteropathogenic, enterotoxigenic, enteroinvasive, and uropathogenic E. coli strains (Fukiya et al. 2004). As is the case for other pathogenic E. coli, it is apparent that the virulent O157:H7 strain has arisen from an ancestral enteropathogenic E. coli O55 by both the acquisition of genetic material containing virulence determinants and the loss of genetic material present in commensal bacteria like the K12 strain (Donnenberg & Whittam 2001). An example of the acquisition of genetic material by E. coli O157:H7 is the pathogenicity island it carries, which is referred to as the LEE (locus for enterocyte effacement) locus that is inserted into the bacterial genome. The low G + C content of the LEE pathogenicity island suggests that acquisition of this virulence cassette originally occurred by horizontal transmission. The LEE contains genes that encode a type three secretion system, which serves as a molecular syringe to deliver bacterial-derived proteins into the cytosol of the host cell to which the bacterium has attached. One of the bacterial proteins, EspE, serves as a receptor for binding of the outer membrane protein intimin, which is encoded by the eae gene also present in the LEE pathogenicity island. A recent report suggests that these effector proteins might serve as effective vaccine candidates to reduce colonization of the gut and decrease fecal shedding of the organism in ruminants (Potter et al. 2004). In animal models and in in vitro tissue culture assays, bacteria containing the LEE locus adhere intimately to the surface of infected cells. Underneath adherent bacteria, the cytoskeleton of the eukaryotic cell is reorganized into an attaching-effacing lesion in which surface microvilli are disrupted and F-actin is recruited into a pedestal directly beneath adherent bacteria. F-actin associated proteins, such as the bridging protein alpha-actinin, are also recruited to the pedestal of the attaching-effacing lesion (Johnson-Henry et al. 2001). Genes coding for putative bacterial attachment factors separate from the LEE pathogenicity island also can be found either elsewhere in the genome or contained on a large plasmid referred to as pO157 (Tarr et al. 2000, Paton et al. 2001, Torres & Kaper 2003). However, the precise role of each of these binding factors during the course of infection in vivo has yet to be defined. Phages that code for toxins have inserted into the genome of enterohemorrhagic E. coli, including O157:H7 strains. These toxins were first identified by their cytopathic effects on Vero cells and were named Verotoxins. Subsequently, O'Brien and her colleagues showed that Verotoxin-1 is highly homologous to the Shiga toxin elaborated by Shigella dysenteriae, type 1 and, therefore, referred to the E. coli-derived toxin as Shiga-like toxin. It is now apparent that there exists a family of closely related Shiga-like toxins. Those associated with human disease bind to the receptor CD77, also referred to a globo-triaosylceramide (Gb3). Shiga-like toxins are multimeric toxins comprised of an active (A) subunit and five binding (B) subunits that attach to Gb3 on the surface of certain, but not all, host cells (O'Loughlin & Robins-Browne 2001). HOST CELL SIGNALING RESPONSES Programmed cell death - In addition to causing cell cytotoxicity, it is now apparent that Shiga-like toxins also induce programmed cell death. Within 24 to 48 h of exposure to the toxin, HEp-2 cells demonstrate hallmark morphologic features of apoptosis, including cell shrinkage, membrane blebbing, and condensation of nuclear chromatin (Jones et al. 2000). These morphologic features of programmed cell death are accompanied by DNA fragmentation, activation of caspases, and degradation of the PARP (poly-ADP-ribose polymerase) deoxyribonucleic acid repair enzyme; features indicative of the activation of an apoptotic response (Ching et al. 2002). Apoptosis of eukaryotic cells depends on the expression of the CD77 receptor, as is evident by the varying susceptibility of B cells to the effects of Shiga-like toxin dependent on the cell cycle expression of Gb3. Induction of apoptotic responses to the toxin in rabbit intestine is developmentally regulated and correlates with expression of CD77. Similarly, Shiga-like toxins do not induce programmed cell death of T84 colonic epithelial cells that do not express detectable levels of CD77. The mechanism of toxin-mediated apoptosis has been under investigation. Activation of the Bcl family of proteins in cells exposed to Shiga-like toxin 1 was demonstrated by immunoblotting of cytosolic proteins. There is evidence of increased levels of the pro-apoptotic family members Bax and Bad, whereas there is a reduction in the amount of the anti-apoptotic Bcl-2 protein. In transient transfection experiments, increased levels of Bcl-2 could prevent the programmed cell death induced by Shiga-like toxin (Jones et al. 2000). In response to exposure of HEp-2 cells to Shiga-like toxin, there is activation of pro-apoptotic caspases, including caspase-3, caspase-8, and caspase-9. Inhibitors of caspase activation reversed these effects (Jones et al. 2000). Activation of Bid to a truncated form (tBid) and release of cytochrome c into the cytosol (Ching et al. 2002) provide complementary evidence for the role of the mitochondrial pathway of programmed cell death in the epithelial response following exposure to the Shiga-like toxins. Current studies are evaluating the role of the apoptotic response to bacterial toxins in vivo by employing relevant animal models. INHIBITION OF STAT-1 SIGNALING The Jak-Stat signaling cascade is involved in mediating host cell cytokine signal transduction, and is also indispensable to the host response to microbial pathogens. A series of seven separate Stat proteins are present in a wide variety of cell types, including cells of epithelial origin (Ihle 2001). Host responses to microbes are mediated through activation of the various Stat proteins. For instance, Stat-1 knockout mice are more susceptible to bacterial infection compared with their wild-type littermates. Furthermore, humans deficient in Stat1 have an increased susceptibility to viruses and mycobacterial infection (Dupuis et al. 2003). Stat-1 mediates a pro-inflammatory response to the activation of the interferon gamma receptor on the cell surface by interferon gamma (Fig. 1). Auto-phosphorylation of Jak proteins attached to the cytoplasmic face of the interferon gamma receptor recruits latent Stat-1 molecules present in the cytosol of the cell. Following dimerization and phosphorylation of specific tyrosine residues, active Stat-1 dimers can then translocate to the nucleus of the cell and bind to a specific DNA sequence referred to as the GAS (for gamma activated sequence), which results in the transcription of a wide array of pro-inflammatory molecules (Meyer et al. 2002). EHEC O157:H7 inhibits the signaling of Stat-1 normally activated by interferon-gamma in epithelial cells (Ceponis et al. 2003). The effects are dependent on viable organisms binding to the cell surface. However, the effects are not mediated by proteins encoded on the LEE locus. For example, STEC of the serotype O113:H21 also inhibits interferon-gamma induced Stat-1 activation, even though the strain contains neither the LEE locus nor causes the attaching-effacing lesion in infected epithelial cells. CONCLUSIONS Microbes, including EHEC, and their products can cause disease in humans by attaching to the cell surface of infected cells, but without subsequent invasion of the organism into the host cytosol. Both the activation (e.g. programmed cell death) and the inhibition (e.g. Stat-1 signaling) of signal transduction responses occur in response to microbial infection (Fig. 2). Interruption of these responses to bacteria and their products could provide novel targets for directing future therapeutic interventions. ACKNOWLEDGEMENTS To Mr Danny Aguilar, Graphics Centre, Hospital for Sick Children for assistance in preparation of the figures contained in this manuscript. REFERENCES
Copyright 2005 Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc05051f2.jpg] [oc05051f1.jpg] |
| |||||||||

{kind=link}
{kind=link}