 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 100, No. 6, October 2005, pp. 575-606 The use of biodiversity as source of new chemical entities against defined molecular targets for treatment of malaria, tuberculosis, and T-cell mediated diseases - A Review Luiz Augusto Basso/********/+, Luiz Hildebrando Pereira da Silva*, Arthur Germano Fett-Neto**, Walter Filgueira de Azevedo Junior********, Ícaro de Souza Moreira***, Mário Sérgio Palma****, João Batista Calixto*****, Spartaco Astolfi Filho******, Ricardo Ribeiro dos Santos*******, Milena Botelho Pereira Soares*******, Diógenes Santiago Santos********/+ Faculdade de Biociências
********Faculdade de Farmácia, Centro de Pesquisas em Biologia Molecular
e Funcional, Financial support: Finep, CNPq, Capes, Instituto do Milênio. All the authors are research fellowship holders of CNPq. Received 20 May
2005 Code Number: oc05132 Corpora non agunt nisi fixata (Paul Ehrlich, 1909) The modern approach to the development of new chemical entities against complex diseases, especially the neglected endemic diseases such as tuberculosis and malaria, is based on the use of defined molecular targets. Among the advantages, this approach allows (i) the search and identification of lead compounds with defined molecular mechanisms against a defined target (e.g. enzymes from defined pathways), (ii) the analysis of a great number of compounds with a favorable cost/benefit ratio, (iii) the development even in the initial stages of compounds with selective toxicity (the fundamental principle of chemotherapy), (iv) the evaluation of plant extracts as well as of pure substances. The current use of such technology, unfortunately, is concentrated in developed countries, especially in the big pharma. This fact contributes in a significant way to hamper the development of innovative new compounds to treat neglected diseases. The large biodiversity within the territory of Brazil puts the country in a strategic position to develop the rational and sustained exploration of new metabolites of therapeutic value. The extension of the country covers a wide range of climates, soil types, and altitudes, providing a unique set of selective pressures for the adaptation of plant life in these scenarios. Chemical diversity is also driven by these forces, in an attempt to best fit the plant communities to the particular abiotic stresses, fauna, and microbes that co-exist with them. Certain areas of vegetation (Amazonian Forest, Atlantic Forest, Araucaria Forest, Cerrado-Brazilian Savanna, and Caatinga) are rich in species and types of environments to be used to search for natural compounds active against tuberculosis, malaria, and chronic-degenerative diseases. The present review describes some strategies to search for natural compounds, whose choice can be based on ethnobotanical and chemotaxonomical studies, and screen for their ability to bind to immobilized drug targets and to inhibit their activities. Molecular cloning, gene knockout, protein expression and purification, N-terminal sequencing, and mass spectrometry are the methods of choice to provide homogeneous drug targets for immobilization by optimized chemical reactions. Plant extract preparations, fractionation of promising plant extracts, propagation protocols and definition of in planta studies to maximize product yield of plant species producing active compounds have to be performed to provide a continuing supply of bioactive materials. Chemical characterization of natural compounds, determination of mode of action by kinetics and other spectroscopic methods (MS, X-ray, NMR), as well as in vitro and in vivo biological assays, chemical derivatization, and structure-activity relationships have to be carried out to provide a thorough knowledge on which to base the search for natural compounds or their derivatives with biological activity. Key words: biodiversity - defined molecular targets - tuberculosis - Apicomplexan - T-cell mediated diseases LESSONS FROM NATURAL MOLECULES Numerous and diverse classes of natural products have been isolated and their structures characterized in the past century. The elucidation of biological and biochemical mechanisms of natural products with therapeutic action have been invaluable to the efforts of organic and medicinal chemists as tools for deciphering the logic of biosynthesis and as platforms for developing front-line drugs (Newman et al. 2000). Natural products and their derivatives have traditionally been the most common source of drugs, and still represent more than 30% of the current pharmaceutical market (Kirkpatrick 2002). Of the 877 small-molecule New Chemical Entities (NCEs) introduced between 1981 and 2002, roughly half (49%) were natural products, semi-synthetic natural product analogues or synthetic compounds based on natural-product pharmacophores, which are an ensemble of steric and electronic features that is necessary to ensure optimal interactions with a specific biological target structure and to trigger or block its biological response (Newman et al. 2003). Natural products are still major sources of innovative therapeutic agents for infectious diseases (both bacterial, parasitic, and fungal), cancer, lipid disorders, and immunomodulation (Altmann 2001). The complexity of many natural products can limit the scope for making chemical modifications to optimize their therapeutical use and can increase the cost of these drugs. Moreover, the need for a renewable supply of active compounds from biological sources can be an obstacle to large-scale production. However, total synthesis in multigram quantities of discodermolide (a potent anti-cancer natural product) shows that the increasing efficiency of synthetic organic chemistry has reduced the barrier posed by limited natural supply, even for compounds with very complex structures (Gunasekera et al. 2004). Moreover, although several important natural products that act with potency and specificity at protein receptors have simple structures (e.g. noradrenalin, adrenaline, serotonin, melatonin, histamine), natural products typically have more stereogenic centres and more architectural complexity than synthetic molecules fashioned by medicinal chemists (Clardy & Walsh 2004). Natural products have taught us about chemical functionality that is compatible with the aqueous milieu of biological microenvironments and built-in conformational constraints of active molecules that reflect the importance of reducing the entropy loss upon ligand binding to biological targets and thus increasing their potency. The functional-group array used by nature has informed synthetic- and medicinal-chemists about bio mimetic strategies and isostere (shape-conserving) replacements. Synthetic molecules are increasingly produced by combinatorial chemistry approaches, in which a common core is elaborated by attaching combinations of fragments to reactive sites on the periphery of the core structure, thereby creating a synthetic combinatorial library. Nature uses similar strategies, especially the oxidative elaboration of a central core followed by capping reactions (methylations, acylations, glycosylation, and oxidations, including hydroxylation). However, even large libraries of compounds from combinatorial chemical approaches used in screening do not reflect the rich chemical diversity indicated by the much larger chemical space covered by natural products (Dobson 2004), since combinatorial libraries are designed more on the basis of chemical accessibility and maximum achievable size than on biologically relevant chemical diversity or properties (Martin & Critchlow 1999). The reason for the lack of lead compounds from synthetic libraries in some therapeutic areas such as anti-infective, immunosuppression, oncology, and metabolic diseases may be due to the different chemical space occupied by natural products and synthetic compounds. Accordingly, combinatorial libraries based on natural product templates, which have been honed by their evolutionary history for biological activity, are excellent starting points for structural diversification to increase the likelihood that a chemical compound with useful biological action is found, especially in therapeutic areas that have a dearth of lead compounds (Brohm et al. 2002). It has long been recognized that natural-product structures have the characteristics of high chemical diversity, biochemical specificity, and other molecular properties that make them favourable as lead structures for drug discovery, and which serve to differentiate them from libraries of synthetic and combinatorial compounds (Clardy & Walsh 2004). Computational chemistry has been used to measure those desirable chemical features that distinguish natural products from other sources of drug leads. Representative combinatorial, synthetic and natural-product compound libraries have been examined on the basis of molecular mass, number of chiral centres, molecular flexibility as measured by number of rotatable bonds and ring topology, distribution of heavy atoms, and Lipinsky-type descriptors (Feher & Schmidt 2003). Cristopher A Lipinski's experimental and computational analyses of the World Drug Index led to the "rule-of-five" that identifies several key properties that should be considered for small molecules that are intended to be orally administered (Lipinski et al. 1997). These properties are: molecular mass < 500 Daltons, number of hydrogen-bond donors < 5, number of hydrogen-bond acceptors < 10, calculated octanol-water partition coefficient (an indication of the ability of a molecule to cross biological membranes) < 5. An analysis of the physical properties and structure fragments of marketed oral drugs concluded that they have lower molecular mass, fewer hydrogen bond acceptors and donors, and fewer rotatable bonds compared with drugs that have other routes of administration (Vieth et al. 2004). Natural products, trade drugs or other synthetic molecular libraries have been differentiated on the basis of scaffold architecture and pharmacophoric properties (Lee & Schneider 2001), or entropy analysis and binary quantitative structure-activity relationship calculations (Stahura et al. 2000). These studies reveal that natural products typically have a greater number of chiral centres and increased steric complexity than either synthetic drugs or combinatorial libraries. Although drug and combinatorial molecules tend to have a significantly higher number of nitrogen-, sulphur- and halogen-containing groups, natural products bear a higher number of oxygen atoms. Natural products differ significantly from synthetic drugs and combinatorial libraries in the ratio of aromatic ring atoms to total heavy atoms (lower in natural products), number of solvated hydrogen-bond donors and acceptors (higher in natural products), and by greater molecular rigidity. Natural-product libraries also have a broader distribution of molecular properties such as molecular mass, octanol-water partition coefficient, and diversity of ring systems compared with synthetic and combinatorial counterparts. Interestingly, the fraction of natural product structures with two or more "rule-of-five" violations is quite low (approximately 10%) and equal to that of trade drugs (Lee & Schneider 2001). Hence these rules can be used to guide selection of natural products derived from plant extracts with drug-like properties to increase the likelihood of discovering a potential therapeutic agent and subsequent market launch of a New Molecular Entity (NME is a medication containing an active ingredient that has not been previously approved for marketing in any form). In addition, natural products have been described as a population of privileged structures selected by evolutionary pressures to interact with a wide variety of proteins and biological targets for specific purposes which is supported by the fact that natural products have become effective drugs in a wide variety of therapeutic indications (Koehn & Carter 2005). For thousands of years medicine and natural products have been closely linked through the use of traditional medicines and natural poisons (Newman et al. 2000). There are a number of sources for natural products that have proved extremely valuable as therapeutic agents: sponges (discodermolide and hemiasterlin), cyanobacteria (apratoxin A and jamaicamide), myxobacteria (epothilone and tubulysins), soil microbes (vancomycin, stauros-porine, rapamycin), dinoflagellates (saxitoxin), and a small-reef dwelling tunicate found in the West Indies (ecteinascidin), to name a few. We will nevertheless focus on a few examples of natural products derived from plants. Clinical, pharmacological, and chemical studies of traditional medicines, which were derived predominantly from plants, were the basis of most early medicines such as aspirin, digitoxin, morphine, quinine, emetin, and pilocarpine. Quinoline antimalarials and related aryl alcohols are based on the chemical structure of quinine, an active ingredient of Cinchona bark, which was first imported into Europe from Peru for antimalarial use in the XVII century by the Jesuits. The dependence on raw material for its extraction and the opportunities presented by its structural elucidation led to the development of fully synthetic and inexpensive 4-aminoquinoline antimalarials - notably chloroquine and amodiaquine (O'Neill et al. 1998). Artemisinin is the active ingredient of the Chinese herb "qinghao" (Artemisia annua) that was traditionally used for treating fevers. Several semi synthetic derivatives of artimisinin have been used increasingly over the past two decades as antimalarials (Haynes 2001). These derivatives include artemether, arteether, and artesunate, which are all metabolized to dihydroartemisinin - the main active agent in the body, are fast acting and act against gametocytes, the sexual stages of Plasmodium that infect mosquitoes. The active ingredient of galantamine (Reminyl) used for treatment of Alzheimer's disease was originally isolated from the plant Galanthus spp. and later from Narcissus spp. Vinblastine is a lead compound for the development of semisynthetic anticancer agents isolated from Catharanthus roseus. Screening of plant extracts against the antimalarial target plasmepsin II identified active compounds from the stem bark and leaves of Albizia adinocephala collected from Panama, and fractionation of the stem bark extract led to isolation of two new macrocyclic spermine alkaloids as the active components (Butler 2004). The primary target of licensed drugs for the treatment of Alzheimer's disease is the inhibition of the enzyme acetylcholinesterase, although preventing β-amyloidosis is a prime target for drugs in development. The in vitro dual anti-cholinesterase and β-secretase activities of Camellia sinensis L. extract (tea) has recently been reported (Okello et al. 2004). However, the active ingredients in tea infusions have not yet been isolated. The phenolic yellow pigment in curry spice, curcumin, may fight Alzheimer's disease, since it has been shown to inhibit the formation of amyloid-b oligomers and fibrils and breaks up amyloid-b plaques in the brains of a murine model (Yang et al. 2005). Human clinical trials of curcumin in patients with mild to moderate cases have begun at UCLA's Alzheimer's Disease Research Center. Clearly, plants hold a prominent position in the available sources of natural bioactive molecules. The sessile character of plants required the development of a large array of molecules to effectively respond to biotic and abiotic stimuli in situ. The chemical diversity of plants has made them the source of choice for the isolation of pharmacologically relevant metabolites. Approximately 25% of the drugs prescribed worldwide come from plants, whereas 11% of the 252 drugs considered as basic and essential by the World Health Organization (WHO) derive exclusively from plants (Rates 2001). Prescription drugs containing phytochemicals were valued at more than US$ 30 billion in 2002 in the US alone (Raskin et al. 2002). Moreover, relatively sophisticated traditional medicine systems (mostly based on plants) have been in existence for thousands of years in countries such as China and India, as well as in the New World tropics (Cragg et al. 1995). According to statistics of the WHO, these systems continue to play the major role in primary health care of approximately 80% of the world's inhabitants (Cragg et al. 1995). Paradoxically, in contrast to this extreme relevance of plants as sources for new drugs, there is a considerable lack of knowledge on plant chemistry compared to the existing diversity, particularly in the tropics. Of the estimated 400,000 higher plant species in the world, only about 10% have been characterized chemically to some extent, whereas of the approximately 100,000 secondary metabolites that have been discovered from the plant kingdom, only half have been fully elucidated (Oksman-Caldentey & Inzé 2004). Therefore, the unexplored potential of plants as a source of novel bioactive chemicals is enormous. Metabolites derived from plants can be used in several different ways in the development of drugs. A natural product can be used intact, as it is isolated from the plant. This is the case of the bisindole alkaloids vincristine and vinblastine, which are isolated from cultivated periwinkle (Catharanthus roseus) plants. These alkaloids induce microtubule disassembly and are used in the treatment of childhood leukemia and Hodgkin's disease. Childhood leukemia victims have a 99% chance of remission, whereas Hodgkin's patients have a 70% chance of cure with the use of these alkaloids (Oksman-Caldentey & Inzé 2004). A precursor of a natural product molecule that is difficult to synthesize and relatively more abundant that the complete molecule can be isolated from the plant and the final assembly is completed through semisynthesis. This is the case of the main form of commercial production of Taxol® (Paclitaxel®), a diterpene amide produced by gymnosperm trees of the genus Taxus. Although taxol can be obtained directly from needles, bark, and cell cultures of Taxus spp. the usual procedure is the isolation of the more abundant (circa 0.1% dw in needles) diterpene precursor 10-deacetyl-baccatin III (10-DAB) and attachment of the easily synthesized N-benzoyl-3-phenylisoserine taxol side chain to the C13 of the 10-DAB skeleton (Jennewein & Croteau 2001). Taxol is an important antitumoral agent increasingly used for the approved treatment of various cancer types, such as ovarian, breast, lung, melanoma, and AIDS-related Karposi's sarcoma (Fett-Neto et al. 2004). The mechanism of action of taxol is the opposite of that of the periwinkle alkaloids, acting as a microtubule stabilizer and blocking cell cycle progression at the G2-M transition (Jennewein & Croteau 2001). A semisynthetic derivative of the natural product can be used as a drug. This is the case of the plant lignan podophyllotoxin, which is highly toxic and isolated from Podophyllum spp. However, semisynthetic derivatives of podophyllotoxin (etoposide®, etopophos® and teniposide®) are important antitumor agents against small cell lung cancer, testicular cancer, acute lymphatic leukemia, and children's brain tumors. The mechanism of action is based on the inhibition of the enzyme topoisomerase II, fundamental in preventing DNA synthesis and replication (Oksman-Caldentey & Inzé 2004). It is also important to note that plant-derived products have served as models for the development of modern drugs, such as, for example, quinine for chloroquine (Kutchan 1995). Many disciplines and resources have to come together to deliver success to drug discovery and development efforts. Genomics can provide many potential molecular targets, but only those targets that can be readily manipulated and tested are likely to provide an opportunity to discover a lead molecule. Accordingly, many potential targets will never be advanced because no chemical lead is identified. When a lead compound is found, chemical synthesis efforts are started to improve enzyme inhibition, and efficacy against the pathogen. However, other factors have to be considered after selecting a molecule for further development such as ease of manufacture (low cost is crucial for antimycobacterials and antimalarials), stability, ready formulation, bioavailability (that is, extensive absorption from the gut and absence of first pass metabolism in the liver to achieve effective concentrations in the systemic circulation), an appropriate half-life, and low toxicity. Good drugs are only useful if they are made available to the people who need them and are used properly. Hence the availability of inexpensive compounds, sound policies on their use, strong healthcare infrastructure, and sufficient funds to purchase the drugs that are needed are all vital (Ridley 2002). In addition, many effective and affordable antimycobacterial and antimalarial drugs should be in reserve to meet the challenge of resistance as it arises. Although the history of chemotherapeutic agent development demonstrates the remarkably successful tinkering of a few structural scaffolds by generations of medicinal chemists to meet the challenges of evolution of drug resistance, it also emphasizes the ongoing, cyclical need for innovation. Although traditionally natural products have played an important role in drug discovery, in the past few years most Big Pharma companies have either terminated or considerably scaled down their natural product operations. This occurs despite a significant number of natural product-derived drugs being ranked in the top 35 worldwide selling ethical drugs in 2000, 2001, and 2002; representing 40% of worldwide drug sales in 2000, 24% in 2001, and 26% in 2002 (Butler 2004). Therefore, in addition to being a proven and important source of drug leads, natural products-derived drugs also contribute significantly to the profitability of many pharmaceutical companies. Despite the many advances in technology, the cost of generating new drugs is inexorably rising, leading to ever greater pressure on pharmaceutical companies to focus on developing therapies primarily for the common diseases of wealthy countries (Service 2004). People in poor countries, particularly in the tropics, are all too often neglected by the pharmaceutical industry in the continuing fight against infections and diseases that most afflict them. ETHNOBOTANY AND ETHNOPHARMACOLOGY The search for new bioactive plant products can follow three main routes: random, ethnobotanical, and ecological search. Random search is considered extremely laborious and yields success rates in the order of 1 new product per 10,000 screened plants. Between 1960 and 1981, the National Cancer Institute (NCI-US) in collaboration with the United States Department of Agriculture (USDA) collected and tested more than 114,000 extracts of some 35,000 plants against a range of animal tumor systems, essentially cell cultures (Cragg et al. 1995). Nonetheless, important drugs have been discovered using this method, such as taxol, derivatives of camptothecin and homoharringtonine. The ethnobotanical approach uses the medical knowledge of traditional societies to select plants for testing bioactivities. The success rates of this approach are substantially higher than those of random screening, with the additional advantage that, to some extent, the continued use of crude preparations are, in fact, comparable to small-scale clinical trials, raising the chances of obtaining something amenable to human use. Tests carried out at the NCI for antineoplastic activity including a subset of plants selected on the basis of traditional medicinal use yielded positive activity in the order of 2 to 5 times higher than random screening (Lewis & Elvin-Lewis 1995). Ecological observation is another way to obtain leads to plant bioactivities. The absence of predation in areas infested with herbivores, for example, can indicate the presence of toxic defense compounds. A variation of this approach is zoopharmacognosy, which proposes the selection of plant species regularly ingested by animals, mostly primates, for reducing pain, microbial or worm infestations (Berry et al. 1995). A literature survey shows that there are plants being used in local communities or in the pharmacopeas of ancient countries (such as China and India) for the treatment of some of the diseases targeted in the current review. Some of these species have displayed positive effects in controlled experiments and are promising leads for the isolation of new bioactive products for treatment. A screening for growth inhibition of Mycobacterium tuberculosis using the microplate Alamar blue assay was carried out with 44 plant species previously chosen on the basis of traditional use against tuberculosis; of these, six species (Chelidonium majus, Pinus brutia, Salvia aethiopsis, Stachys sylvatica, Ulmus glabra, Urtica dioica) showed activity, the last three in the water fraction (Tosun et al. 2004). Some of these taxa or closely related ones are represented in the Brazilian ecosystems. Another study carried out with 22 Mexican plants traditionally used for treating respiratory diseases showed effective inhibition of multi-drug resistant forms of M. tuberculosis (Jimenez-Arellanes et al. 2003). Extracts were tested against monoresistant variants of M. tuberculosis H37Rv (isoniazid, rifampin, streptomycin, ethambutol resistant) and the hexane extract of Lantana hispida showed the best activity. Remarkably, L. hispida hexane extract was also active against a group of MDR-TB clinical isolates but did not inhibit the growth of non-tuberculous mycobacteria. The hexane extract of L. hispida was fractionated by column chromatography and one of its fractions (FVI) inhibited the growth of all the MDR-TB clinical isolates at concentrations up to 25 mg ml-1. This study further supports the fact that selecting plants by ethnobotanical criteria enhances the probability of finding active species. The genus Lantana is well represented in the Brazilian ecosystems and could be screened for active compounds. A large array of plants has been reported as having antimalarial activity to various extents. In Brazil, an investigation on the antiplasmodial activity of three species used as antimalarial indicated activity in bark extracts of Remijia ferruginea, an alkaloid producing species but devoid of quinine (Andrade-Neto et al. 2003). Significantly, extracts of a number of species have been shown to display antiplasmodium activity at various concentrations against choroquine-resistant lines of Plasmodium falciparum. Tests also carried out in Brazil with Bidens pilosa (Asteraceae) extracts were very encouraging (Andrade-Neto et al. 2004). The extracts were active in mice infected with P. berghei: doses of up to 500 mg kg-1 administered by the oral route reduced malaria parasitaemia and mouse mortality. Tests in vitro against three P. falciparum isolates, two chloroquine resistant and one mefloquine resistant, showed activity of plants grown in a number of conditions, but higher activities were observed in wild plants. Analysis using thin layer chromatography displayed flavonoids (presumably responsible for the antimalarial activity) in all plants tested, even though at different profiles. According to the authors, because B. pilosa is proven to be active against P. falciparum drug-resistant parasites in vitro, and in rodent malaria in vivo, it is a good candidate for pre-clinical tests as a phytothera-peutic agent or for chemical isolation of the active compound(s) with the aim of finding new antimalarial drugs. Studies with plants traditionally used for malaria treatment from various parts of the world (Vietnam, South Africa, and São Tomé and Príncipe) also showed inhibitory activities against chloroquine sensitive or resistant strains of P. falciparum (Tran et al. 2003, Nundkumar & Ojewole 2002). Particularly promising results were obtained with extracts of the following species: Coscinium fenestra (whole plant), Psidium guajava (bark), Vangueria infausta (leaf), Struchium spargano-phorum (leaf), Cinchona succirubra (bark), Tithonia diversifolia (shoots), Cedrela odorata (bark), and Pycnanthus angolensis (bark). Once again, related taxa or even some of the same species are available in the Brazilian ecosystems. Plants are also a major source of traditional medicines and chemicals with useful properties in the modulation of the immune system. Traditional herbal preparations from the Ayurveda, particularly one containing Alpinia calcarata, have yielded significant improvements in patients suffering from rheumatoid arthritis (Thabrew et al. 2001). Physalis angulata, an annual herb widely used in folk medicine as treatment of various illnesses including rheumatism and asthma, has a series of seco-steroids (physalins) with potent immunomodulatory activity (Soares et al. 2003). Traditional medicine provides a potential source of new plant-derived therapeutic agents against psoriasis (Amenta et al. 2000). Moreover, a traditional herbal medicine from Korea used to treat allergic diseases, containing eight plant species, has been shown to modulate T cell activity (Ko et al. 2004). Therefore, there are several leads available for the search of plant-derived compounds with immune-modulatory activity. A key feature in phytochemical analysis is the ocurrence of certain groups of secondary metabolites in botanical families or subfamilies. This has been the basis of chemotaxonomy, a tool that has been assisting taxonomists in the resolution of certain taxa in conjuction with more powerful methods such as molecular taxonomy (Wink 2003). The fact that certain taxa are more prone to contain biochemical pathways leading to particular groups of secondary products can be successfully used to select plant species for screening bioactivities. A combined approach of ethnobotanical and chemotaxonomical leads has unveiled a number of bioactive species from the genus Psychotria in Brazil, resulting in the discovery of new alkaloids with interesting pharmacological properties (Elisabetsky et al. 1997, Both et al. 2002, Gregianini et al. 2003). PROSPECTION OF BRAZILIAN BIODIVERSITY Species diversity is not evenly distributed across the planet. Approximately 70% of the world's species occur in only 12 countries: Australia, Brazil, China, Colombia, Ecuador, India, Indonesia, Madagascar, Mexico, Peru, and Zaire. In addition, the tropical rainforests of the world are believed to contain more than half the number of species on earth. Brazil has a prominent position in the world's biodiversity, since it contains two extensive areas regarded as biodiversity hotspots, the Atlantic Forest in the Coast and the Cerrado in Central Brazil. A biodiversity hotspot is defined as the area that contains at least 0.5% of the world's 300,000 known plant species as endemics (Myers et al. 2000). The large biodiversity within the territory of Brazil puts the country in a strategic position to develop the rational and sustained exploration of new metabolites of therapeutic value. The extension of the country, particularly in the North-South direction, but also in the East-West, covers a wide range of climates, soil types and altitudes, providing a unique set of selective pressures for the adaptation of plant life in these diverse scenarios. Chemical diversity is also driven by these forces, in an attempt to best fit the plant communities to the particular abiotic stresses, fauna, and microbes that co-exist with them. The areas of plant sampling proposed in the present study (Amazonian Forest, Atlantic Forest, Araucaria Forest, Caatinga) are rich in species and types of environments. They span different climates, soils, and altitudes, providing a unique set of environmental variables for the specialization of plants and development of chemical defenses. The Amazonian Forest is well known for its diversity of species, many of which unknown to science. Several physionomical elements have been recognized in the Amazonian Forest. For simplicity, the focus must be on the areas of proper forest, excluding the savana-like regions, fields, and the palm-tree rich areas that also occur in the Amazonian region. The forest areas can be divided into Dry Land Forest ("Mata de Terra Firme"), Transiently Flooded Forest ("Mata de Várzeas"), and Permanently Flooded Forest ("Mata de Igapó") (Ferri 1980). The Dry Land Forest is distant from rivers and located in higher parts of the landscape. This Forest is permanently green, with little and non-synchronized change of foliage. Density of trees is rather high and leaves are mostly broad. Large trees characterize this forest, such as Bertholletia excelsa (castanheira - Brazil nut) and Castilloa ullei (caucho). The tree canopy is often continuous, forming a dense obstacle to light, retaining up to 90% of the incident irradiance. Hence, the forest floor is dark, poorly ventilated, and humid. The forest is rich in members of Leguminosae, Moraceae, and Sapotaceae. Palm tree species, such as Astrocaryum, Attalea, and Geonoma, are also frequent. Triplaris and Tococa, plants that associate with ants, are also represented. In the understorey Araceae, Gesneriaceae, Piperaceae, Bromeliaceae, and ferns predominate. Transiently Flooded Forest has variable composition, containing elements of the other two types of Forest of the Amazonian region. Species of Moraceae, Sapo-taceae, and Leguminosae grow into large trees, forming a large part of the canopy. Important species in this Forest are Calycophyllum spruceanum, Dipteryx odorata (the coumarin producing Tonka bear tree), Hevea brasiliensis (rubber tree), Dinizia excelsa, Andira, Parkia, Hymenaea (Ferri 1980). The Permanently Flooded Forest components include relatively smaller trees (up to 20 m) often producing large support roots and stem perforations (lenticels) for aeration. Typical trees include Triplaris surinamensis, Bombax aquaticum, and Macrolobium acaciaefolium. Many epiphytes are represented in this area, including Piperaceae, Araceae, Orchidaceae, Bromeliaceae, Cactaceae, Gesneriaceae, ferns, mosses, and liverworts. The Atlantic Forest is characterized by a high biodiversity comparable to that of the Amazon Forest. The proximity to the Atlantic ocean provides a stable source of humidity, allowing high vegetation density. Trees are dominant, which may reach 30 m in height; there are numerous liana and epiphytes. The shaded and poorly ventilated understorey contains many herbaceous species. A floristic survey of the southern limit of Atlantic Forest area has been carried out, revealing many species of economic interest (Ferri 1980), many of which with medicinal properties. Among the upper storey tree species, the "canelas" (Ocotea sp.) and other Lauraceae, are valued for their wood and aromatic essences; the "ipes" (Tabebuia sp., Bignoniaceae, pau d'arco), appreciated for their wood and phytomedicinal uses; Euterpe edulis, the "palmito", from which palm hearts are extracted; the Cecropia and Inga, two genera that establish associations with ants. Epiphyte species include members of Araceae, Gesneriaceae, Piperaceae, Moraceae, and Orchidaceae and Bromeliaceae of high ornamental value. Lower storey trees include Psychotria umbellata, which produces psy-chollatine (formerly known as umbellatine), a novel monoterpene indole alkaloid with morphine-like analgesic activity (Paranhos et al. 2005). At the forest border, members of Leguminosae can be found, many of which often accumulate toxic proteins, amino acids, and alkaloids. Herbaceous species include members of families known for bioactive metabolites (e.g., alkaloids, terpenes, phenolics, peptides, proteins), such as Apocynaceae, Solanaceae, Ery-throxylaceae, Lamiaceae, Asteraceae, Leguminosae, and Euphorbiaceae. The herbaceous layer is also rich in Pteridophytes and Bryophytes, some of which have antimicrobial compounds. The Araucaria Forest is characterized by a relatively regular distribution of rain fall throughout the year in contrast to most other parts of the country, particularly the Western-Central and Northeast regions. Wide difference between seasons with distinct temperatures and photoperiods are another feature that is peculiar for Brazilian standards. The occurrence of frost is frequent and snowfall can also happen occasionally. Soil conditions also vary, with predominance of basalt-derived purple soils, very rich in nutrients, and stretches of granitic soils much less fertile. As the name suggests, the Araucaria Forest is rich in the Brazilian Pine, Araucaria angustifolia. Other notable components of the forest include: Actinostemom concolor, Sorocea ilicifolia, Urera baccifera, Phytolacca dioica, Cedrela fissilis, Myrocarpus frondosus, Enterolobium contortisiliqum, and species of Sloanea, Ocotea, and Nectandra (Ferri 1980). Another important gymnosperm in this forest is Podocarpus lambertii. Taxol has been reported in needles of Podocarpus gracilior, the first non-taxaceae to yield this chemical (Stahlhut et al. 1999). Among the tree species worth mentioning are also Symplocos uniflora, Ocotea pulchella, Rapanea venosa, Feijoa sellowiana, Eugenia uniflora and associated epiphytes, such as Polypodium sp. (Pteridophytae), and Tillandsia tenuifolia (Bromeliaceae). The Caatinga region of the Brazilian Northeast is characterized by high temperatures, relatively low average humidity, and annual rainfall around 250 to 500 mm. The dry season lasts seven months or more. The so called winter is the rainy season, in which temperatures are not as high. The soils are of different origins and, as a rule, are chemically fertile, well drained, and oxygenated. Water bodies are rarely permanent, drying completely during the summer. Among the dominant species of the Caatinga, one can mention: Amburana cearensis (Leguminosae), Aspidosperma pyrifolium (Apocynaceae), Caesalpinia pyramidalis, Caesalpinia microphylla (Leguminosae), Senna uniflora (Leguminosae), Croton campestris, Euphorbia phosphorea (Euphorbiaceae), Maytenus rigida (Celastraceae), Mimosa caesalpinifolia (Leguminosae), Nicotiana glauca (Solanaceae), Sapium cicatricosum (Euphorbiaceae), and Tabebuia caraiba (Bignoniaceae). The basis for sustained exploration of biodiversity relies on clonal propagation and seed banks for preservation of genetic diversity. If an extract of a determined plant strongly binds to an enzymatic target, it is important to immediately initiate studies on the propagation of the species to supply biomass for extraction, fractionation and isolation of the active metabolite(s). As was shown by the development of taxol as a drug, an early start on the production of catalytically active biomass of source plant species is important to supply material for pre-clinical and clinical tests, as well as for eventual therapeutic use (Cragg et al. 1995). One of the fastest ways of developing a plant species as a sustainable new source for a drug is clonal propagation. The identification of high-yielding genotypes and their mass clonal propagation can significantly enhance drug supply. Clonal propagation can be achieved by different methods, such as rooting of cuttings, development of minicutting propagation programs, in vitro shoot propagation and rooting of microcuttings and somatic embryogenesis. The in vitro methods, in spite of being relatively more expensive, provide a unique opportunity to evaluate the contribution of plant associated microorganisms to the production of a particular bioactive metabolite. This is important because it allows the establishment of the biochemical nature of the active compounds, i.e. if they are genuine plant products derived from a microorganism metabolism or from a plant's response to microbial presence. The relevance of microbial association with plants is highlighted by the widespread occurrence and abundance of endophytes and mycorrhizae in practically all known plant species, particularly in the tropics. These microorganisms do not impart any clear damage to the plants that grow under normal conditions and, on the contrary, provide a series of advantages for the host plant, such as better mineral nutrition and water supply (mycorrhizae) and protection against pathogenic microbes and higher competitive ability with other plant species (endophytes) (Van der Heijden et al. 1998, Scott 2001). The maintenance of the genetic variability within a species of interest is also relevant. Seed propagation is a way to preserve such genetic diversity. As a plant species is progressively "domesticated" for the purpose of obtaining pharmacologically active chemicals, there may be a need for rescuing genes such as those for disease resistance or that confer a particular plant architecture. By keeping a pool of individuals sexually propagated from the natural populations, this reservoir of genetic variability can remain available. TUBERCULOSIS (TB) TB remains the leading cause of mortality due to a bacterial pathogen, M. tuberculosis. The interruption of centuries of decline in case rates of TB occurred, in most cases, in the late 1980s and involved the US and some European countries due to increased poverty in urban settings and the immigration from TB high-burden countries (Raviglione 2003). Thus, no sustainable control of TB epidemics can be reached in any country without properly addressing the global epidemic. It is estimated that 8.2 million new TB cases occurred worldwide in the year 2000, with approximately 1.8 million deaths in the same year, and more than 95% of these were in developing countries (Corbett et al. 2003). In addition, approximately 12% (226,000) of total deaths from TB was attributed to co-infection with M. tuberculosis and human immuno-deficiency virus (TB-HIV). Immune deficient patients with HIV are at increased risk of latent M. tuberculosis infections progressing to active disease, and being transmitted to others (Morens et al. 2004). Approximately 2 billion individuals are believed to harbor latent TB based on tuberculin skin test surveys (Dye et al. 1999), which represents a considerable reservoir of bacilli. It has been estimated that 3.2% of the world's new cases of TB, in 2000, were multidrug-resistant tuberculosis (MDR-TB), defined as resistant to at least isoniazid and rifampicin (Espinal 2003). According to the 2004 Global TB Control Report of the WHO, there are 300,000 new cases per year of MDR-TB worldwide, and 79% of MDR-TB cases are now "super strains", resistant to at least three of the four main drugs used to treat TB (WHO 2004). Localized high incidence rates of MDR-TB have been found in particular regions, which have been defined as hot zones based on either areas where the prevalence of MDR-TB cases is > 5% (that is, where > 5% of current cases are MDR-TB) or areas where the incidence of MDR-TB cases is > 5% (that is, where > 5% of new cases are MDR-TB) (Espinal 2003). MDR-TB is an airborne bacterium that is spread just as easily as drug-sensitive TB. An individual who is sick with any strain of TB will infect between 10 and 20 people each year with that same strain (Nunn 1997), thereby making the hot zones of particular concern to public health officials. More recently, a mathematical model using Monte Carlo sampling methods has been employed to determine the key causal factors in generating hot zones (Blower & Chou 2004). Paradoxically, areas with programs that successfully reduced wild-type pansensitive strains (as a result of high case detection and treatment rates) often evolved into hot zones. It has therefore been suggested that second-line drugs be quickly introduced to disrupt the amplification of resistance. However, the bacteriostatic second-line drugs [amikacin, kanamycin, capreomycin, cycloserine, para-aminosalicylic acid (PAS), ethionamide, fluoroquinolones] are more toxic and less effective and are given for at least three times as long and at 100 times the cost of basic short-course chemotherapy regiments (McKinney et al. 1998, Pablos-Méndez et al. 2002). The factors that most influence the emergence of drug-resistant strains include inappropriate treatment regimens, and patient noncompliance in completing the prescribed courses of therapy due to the lengthy standard "short-course" treatment or when the side effects become unbearable (Duncan 2003). TB is transmitted by M. tuberculosis-containing aerosolized droplets generated by the cough of a person infected with bacilli. The primary infection involves replication of M. tuberculosis within alveolar macrophages of the terminal air spaces of the lung, spread to local lymph nodes within the lung, and eventual dissemination of infection to remote sites in the body within one or two years after an initial infection. Only 5-10% of infected individuals develop the disease because the host immune response against M. tuberculosis is highly effective in controlling bacterial replication. However, M. tuberculosis is almost never eradicated due to its ability to establish and maintain latency, a period during which the infected person does not have clinically apparent TB. Post-primary TB, which is predominantly a pulmonary disease, develops later in life, and can be caused either by reactivation of bacteria remaining from the initial infection or by failure to control a subsequent infection. Post-primary TB involves extensive damage to the lungs and efficient aerosol transmission of bacilli. Effective chemotherapies that decreased TB mortality rate worldwide was led by the discovery of the antibacterial and antitubercular properties of streptomycin in 1944 (Schatz et al. 1944), and both isoniazid and pyrazinamide in 1952 (Middlebrook 1952, Kushner et al. 1952). The later introduction of ethionamide, rifampicin, ethambutol, and ciprofloxacin to the arsenal used to treat tuberculosis seemed to provide an adequate number of effective antimicrobial agents (Basso & Blanchard 1998). Effective tuberculosis chemotherapy must include early bactericidal action against rapidly growing organisms and subsequent sterilization of the semidormant and dormant populations of bacilli (Mitchison 1985). The first-line drugs isoniazid, rifampicin, streptomycin, and ethambutol exhibit early bactericidal activity against actively metabolizing bacilli (Heifets 1994). Pyrazinamide is active against the semi-dormant bacilli in acidic intracellular environments. The modern, standard "short-course" therapy (Mitchison 1985) for TB is based on a four-drug regimen of isoniazid, rifampicin, pyrazinamide, and ethambutol or streptomycin for two months, followed by treatment with a combination of isoniazid and rifampicin for an additional four months. This combination therapy must be strictly followed to prevent drug resistance and relapse, and direct observation of patient compliance is the most reliable way to ensure effective treatment and prevent the acquisition of resistance. The DNA mutations identified in drug-resistant strains of M. tuberculosis have been reviewed (Basso & Blanchard 1998, Ramaswamy & Musser 1998, Glickman & Jacobs 2001, Schroeder et al. 2002). The bacteriostatic second-line drugs ethionamide, cycloserine, and PAS are reserved to strengthen the treatment of drug-resistant disease or when bactericidal drugs are prohibited because of toxicity (Dutt & Stead 1994). M. tuberculosis has been considered the world's most successful pathogen and this is largely due to the ability of the bacillum to persist in host tissues, where drugs that are rapidly bactericidal in vitro require prolonged administration to achieve comparable effects (Hingley-Wilson et al. 2003, Gomez & McKinney 2004). Hence more effective and less toxic anti-tubercular agents are urgently needed to shorten the duration of current treatment, improve the treatment of MDR-TB, and to provide effective treatment of latent tuberculosis infection (O'Brien & Nunn 2001). VALIDATED TARGETS FOR ANTI-TB DRUG DEVELOPMENT A promising target for drug development should be essential for survival of the human pathogen and absent from the human host, which should hopefully result in the development of non-toxic therapeutic agents to treat infectious diseases. Below we briefly describe validated targets for anti-TB drug development of two biosynthetic pathways in M. tuberculosis. BIOSYNTHEIS OF MYCOLIC ACIDS (FAS-II SYSTEM) The mycobacterial cell wall is comprised of three covalently linked macromolecules: peptidoglycan, arabinogalactan, and mycolic acid, which is often described as mycolyl-arabinogalactan-peptidoglycan complex (mAGP; Fig. 1) (Brennan & Nikaido 1995, Schroeder et al. 2002, Brennan 2003). Mycolic acids have become one of the defining taxonomic characteristics of many species in genera such as Mycobacterium, Corynebacterium, Dietzia, Nocardia, Rhodococcus, and Tsukamurella. Mycolic acids are high-molecular-weight a-alkyl, β-hydroxy fatty acids (Fig. 2), which appear mostly as bound esters in tetramycolylpentaarabinosyl clusters in the mycobacterial cell wall. In the pyrolytic cleavage of mycolic acids the intact fatty acid released is often referred to as a branch since it occupies the alpha position with respect to the carboxylic acid group. The aldehyde released is referred to as the meroaldehyde and the corresponding segment of the intact mycolate is often referred to as the meromycolate branch. Isoniazid (INH, isonicotinic acid hydrazide; Fig. 3) is one of the oldest synthetic antitubercular compounds, and the most prescribed drug for active infection and prophylaxis (Blanchard 1996). The product of the M. tuberculosis inhA structural gene (InhA or ENR), which is located downstream of an ORF coding for a β-ketoacyl reductase (MabA) in the inhA operon, has been shown to be a major target for isoniazid (Banerjee et al. 1994). InhA was identified as an NADH-dependent enoyl-ACP (acyl carrier protein) reductase enzyme, which exhibits specificity for long-chain (C18 > C16) enoyl thioester substrates (Quémard et al. 1995). InhA is a member of the mycobacterial Type II dissociated fatty acid biosynthesis system (FASII), which elongates acyl fatty acid precursors yielding the long carbon chain of the meromycolate branch of mycolic acids, the hallmark of mycobacteria (Schroeder et al. 2002). Consistent with InhA as the major target of INH mode of action, inactivation of the M. tuberculosis inhA-encoded enoyl reductase and INH treatment resulted in similar morphological changes to the mycobacterial cell wall leading to cell lysis (Vilchèze et al. 2000). Overexpression of inhA has been shown to confer resistance to INH and ethionamide (ETH) in M. smegmatis, M. bovis BCG, and M. tuberculosis (Larsen et al. 2002). Further biochemical and genetic evidence has been given likewise showing that InhA is the primary target of INH (Kremer et al. 2003). Consistent with these results, mutations in the inhA structural gene and in the inhA locus promoter region have been associated with isoniazid resistance (Basso et al. 1998). Moreover, INH-resistant clinical isolates of M. tuberculosis harboring inhA-structural gene missense mutations, but lacking mutations in the inhA promoter region, katG gene and oxyR-ahpC region, were shown to have higher dissociation constant values for NADH than wild-type (WT) InhA, whereas there were only modest differences in the steady-state parameters (Basso et al. 1998). Consistent with these results, a comparison of the crystal structure of a binary complex of WT and INH-resistant mutant enoyl reductases with NADH (Dessen et al. 1995) showed that disruption of a hydrogen bond network in the mutant protein could account for the higher dissociation constant value for the coenzyme. More recently, molecular dynamics simulations of WT and INH-resistant clinical isolates of M. tuberculosis showed that mutations of the glycine-rich loop residues I21V and I16T resulted in a change in the pattern of direct hydrogen bond contacts with the pyrophosphate moiety of NADH, in which the pyrophosphate moiety of NADH moves apart from its binding site (Schroeder et al. 2005). The increase in the NADH dissociation constant observed for the I21V and I16T isoniazid-resistant mutants (Basso et al. 1998) can be attributed to a decrease in the number of direct hydrogen bond interactions between the cofactor and aminoacids in the binding pocket of the enzyme and to a decrease in water-mediated interactions between cofactor and enzyme (Schroeder et al. 2005). The correlation between the NADH binding properties in solution, molecular dynamics simulation studies and resistance to INH inactivation of M. tuberculosis InhA mutants provides a mechanism of resistance at molecular level for this clinically important drug. The mechanism of action of isoniazid is complex, as mutations in at least five different genes (katG, inhA, ahpC, kasA, ndh) have been found to correlate with isoniazid resistance (Schroeder et al. 2002). However, a drug target for a bactericidal drug has been suggested to be an enzyme of the bacterium (i) that binds the drug, (ii) that is inhibited by the drug, and (iii) whose inhibition induces the death of the bacterium (Vilchèze et al. 2000). Accordingly, based on these criteria, genetic and biochemical results point to InhA as the bona fide target for INH in M. tuberculosis. Deletions of, or missense mutations in, the katG gene have been associated with decreased susceptibility to INH in approximately 50% of clinical isolates of M. tuberculosis (Heym et al. 1995, Ramaswamy et al. 2003). It has recently been shown that the S315T KatG mutant, which is one of the most commonly encountered substitutions in clinical INH-resistant strains, has reduced affinity for INH, which would result in decreased drug activation and ensuing INH resistance (Yu et al. 2003). INH is a pro-drug that is activated by the mycobacterial katG-encoded catalase-peroxidase enzyme in the presence of manganese ions, NAD and oxygen (Johnsson & Schultz 1994, Johnsson et al. 1995, Basso et al. 1996, Zabinski & Blanchard 1997). The KatG-produced acylpyridine fragment of isoniazid is covalently attached to the C4 position of NADH (Fig. 3), and forms a binary complex with the wild-type enoyl reductase of M. tuberculosis (Rozwarski et al. 1998). This isonicotinyl-NAD+ adduct has been characterized spectroscopically and shown to bind to InhA with a dissociation constant value lower than 0.4 nM (Lei et al. 2000). The isonicotinyl-NAD+ adduct has recently been shown to be a slow, tight-binding competitive inhibitor of WT InhA (Rawat et al. 2003). The initial rapidly reversible weak binding (Ki = 16 nM) is followed by a slow isomerization leading to a tighter enzyme-inhibitor complex with an overall dissociation constant (Ki*) value of 0.75 nM. The isoniazid mechanism of action requires the conversion of INH by mycobacterial KatG into a number of electrophilic intermediates (Johnsson & Schultz 1994). It has been suggested that formation of the isonicotinyl-NAD+ adduct may be through addition of either an isonicotinic acyl anion to NAD+ or an isonicotinic acyl radical to an NAD free radical (Rozwarski et al. 1998). More recently, an enzyme-catalyzed mechanism for INH activation has been proposed, leading to isonicotinoyl radical formation (thought to be the activated form of INH) and production of the amide end product via a diazene intermediate (Pierattelli et al. 2004). In the proposed mechanism, the oxyferryl group of compound I of KatG, generated after reaction with peroxide, is reduced by INH in a single electron transfer to the heme with concomitant transfer of a proton from the hydrazide moiety to His-108. The C-N bond of hydrazide is broken, yielding a diazene and the acyl radical. The diazene intermediate (that may be stabilized by Trp107, Asp137 or the oxyferryl group) is reduced to hydrazine and ammonia, which may involve deprotonation of His108, Asp137 or Arg104 of KatG. The serendipitous discovery of isoniazid has now provided clues into critical and unique biosynthetic pathways in mycobacteria. The chemical simplicity of this molecule, and decades of synthetic endeavors after the initial discovery of INH, suggest that more potent analogs, prepared by classic organic synthesis or novel combinatorial synthetic methods, would appear to be hard to find. However, given the oral availability and favorable toxicology profile, INH analogs appear to be worthy of examination as antitubercular agents. Accordingly, based on the mechanism of activation proposed for isoniazid, via electron transfer reaction (DeBarber et al. 2000), an alternative self-activation route has recently been proposed (Oliveira et al. 2004) for designing new drugs for the treatment of WT and INH-resistant tuberculosis. These drugs would be activated by electron transfer reactions before interacting with their cellular target. Most of the INH resistance is associated with katG structural gene alterations resulting in catalase-peroxidase mutant enzymes with impaired ability to form activated-INH intermediates. One approach to overcome this drug resistance is based on the synthesis of a new molecule capable of promoting an inner-sphere electron transfer reaction. In this context, the use of a redox reversible metal complex coordinated to the pro-drug appears as a very first system. Accordingly, we have recently shown that a pentacyano (isoniazid) ferrateII complex inhibits enzyme activity of both wild-type InhA and I21V mutant InhA identified in isoniazid-resistant clinical isolates of M. tuberculosis (Oliveira et al. 2004). The in vitro kinetics of inactivation indicate that this process requires no activation by KatG, no need for the presence of NADH, and is also effective against INH-resistant mutant InhA. An MIC value of 0.2 mg ml-1 for this inorganic complex was determined by the radiometric BACTEC AFB system for M. tuberculosis H37Rv strain, and toxicity assays in HL60 leukemia and MCS-7 breast cancer cells yielded an IC50 value > 25 mg ml-1; thereby indicating a good selectivity index (SI = IC50/MIC > 125; as suggested by the Tuberculosis Antimicrobial Acquisition & Coordinating Facility of US for a compound to move forward through screening programs SI should be larger than 10). More recently, we have shown that the pentacyano(isoniazid)ferrateII complex is a slow-onset inhibitor of M. tuberculosis InhA enzyme activity, with a true overall dissociation constant value of 70 nM (Oliveira et al. 2005). In this mechanism of action an initial enzyme-inhibitor complex is rapidly formed, which then undergoes a slow isomerization reaction to an enzyme-inhibitor binary complex in which the inhibitor is more tightly bound to enzyme. The weakness in the use of classical enzyme inhibitors as drugs for clinical conditions is that inhibition results in the upstream accumulation of the substrate for the enzyme, which may overcome the inhibition. By contrast, the build up of substrate cannot have any effect on the isomerization of enzyme-inhibitor complex typical of the slow-onset mechanism and hence reversal of the inhibition (Morrison & Walsh 1988). In addition, a half-time value of 630 min (10.5 h) for the limiting step for inhibitor dissociation from the binary complex is a desirable feature since it may be expected to enhance inhibitor's effectiveness (Schloss 1988). This inorganic complex may represent a new class of lead compounds to the development of anti-tubercular agents aiming at inhibition of a validated target and effective against isoniazid-resistant strains. Fatty acid elongation occurs through repetitive cycles of condensation, β-keto reduction, dehydration, and enoyl reduction which are catalyzed by, respectively, β-ketoacyl synthase (KAS, condensing enzyme), β-ketoacyl reductase (KAR), β-hydroxyacyl dehydrase (DE), and enoyl reductase (ENR). These chemical reactions are catalyzed by two types of fatty acid synthase systems (FAS). The FAS-I system is a multidomain polyprotein that encodes all the enzymes necessary for fatty acid synthesis in one large polypeptide and is generally present in most eukaryotes, except in plants (Cronan & Rock, 1996). FAS-II systems, which are present in bacteria and plants, catalyze the individual reactions by separate proteins readily purified independently of the other enzymes of the pathway and are encoded by unique genes. Mycobacteria, unlike most organisms, have both FAS-I and FAS-II systems (Brindley et al. 1969). The mycobacterial FAS-I system catalyzes not only the synthesis of C16 and C18 fatty acids, the normal products of de novo synthesis, but also elongation to produce C24 and C26 fatty acids (Bloch 1975). The Type II fatty acid synthase of mycobacteria is analogous to other bacterial FAS-II systems, with the notable exception of primer specificity. The mycobacterial FAS-II is not capable of de novo synthesis from acetate but instead elongates palmitoyl-ACP to fatty acids ranging from 24 to 56 carbons in length (Mdluli et al. 1998, Slayden & Barry 2002). In summary, the mycobacterial FAS-I produces a bimodal (C14:0-C16:0 to C24:0-C26:0) distribution of acyl-CoA fatty acids. The mycobacterial FAS-I system would provide the shorter acyl-CoA fatty acid precursors (C14:0-C16:0) for condensation with malonyl-ACP by mtFabH enzyme activity whose products, in turn, would be elongated by the FAS-II system, yielding the long carbon chain of the meromycolate branch (50-60 carbons) of mycolic acids. The longer chain acyl-CoA products (C24:0-C26:0) of FAS-I would be excluded from chain elongation and remain available to be utilized, presumably in the CoA form, as substrates for formation of the a-alkyl branch (20-26 carbons) of mycolic acids. The complete genome sequence of M. tuberculosis H37Rv reveals the organization of the genes encoding the components of the FAS-II system (Cole et al. 1998). In particular, in the inhA operon, the inhA gene that codes for a trans Δ2-enoyl reductase (InhA - the target for isoniazid mode of action) is located immediately downstream of the mabA (also fabG1 in M. tuberculosis or fabG in Escherichia coli) gene that codes for a β-ketoacyl reductase (MabA) (Banerjee et al. 1998). MabA has recently been shown to preferentially metabolize long-chain fatty acids, consistent with its role in elongation of mycolic acid precursors (Marrakchi et al. 2002). Thus the M. tuberculosis mabA-encoded NADPH-dependent β-ketoacyl reductase enzyme may also represent a target for anti-TB agent development. Accordingly, MabA activity has been shown to be essential for viability of M. tuberculosis by knockout of the mabA gene (Dr Tanya Parish, pers. commun.). THE SHIKIMATE PATHWAY The shikimate pathway is an attractive target for the development of herbicides and antimicrobial agents because it is essential in algae, higher plants, bacteria and fungi, but absent from mammals (Fig. 4; Bentley 1990, Herrmann & Weaver 1999, Coggins et al. 2003). The shikimate pathway links metabolism of carbohydrates to biosynthesis of aromatic compounds. The first committed step in the shikimate pathway is the stereospecific condensation of phosphoenolpyruvate (PEP) and D-erythrose 4-phosphate (E4P) forming 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) and inorganic phosphate catalyzed by DAHP synthase (DAHPS; EC 4.1.2.15) (Fig. 4). The second reaction is the elimination of phosphate from DAHP to generate 3-dehydroquinate (DHQ). The reaction is catalyzed by DHQ synthase (DHQS; EC 4.6.1.3) and requires catalytic amounts of NAD+ for activity, even though the enzyme catalyzed reaction is redox neutral. In the third enzyme-catalyzed reaction, DHQ is dehydrated to 3-dehydroshikimate (DHS) by 3-dehydroquinate dehydratase (often referred to as 3-dehydroquinase; DHQ dehydratase; EC 4.2.1.10), thereby initiating the process of aromatization by introducing the first of three double bonds. The type II enzyme catalyzes a trans-dehydration via an enolate intermediate as in M. tuberculosis, whereas the type I catalyzes a cis-dehydration of 3-dehydroquinate via a covalent imine intermediate as in E. coli. The fourth is the reduction of 3-dehydroshikimate to shikimate catalyzed by shikimate dehydrogenase (aroE-encoded SHK dehydrogenase; EC 1.1.1.25). Shikimate kinase (SK; EC 2.7.1.71), the fifth enzyme of the pathway, catalyzes a phosphate transfer from ATP to the carbon-3 hydroxyl group of shikimate forming shikimate 3-phosphate (S3P). The sixth reaction is an unusual transfer of an enolpyruvyl moiety from phosphoenolpyruvate (PEP) to the 5-hydroxyl group of S3P with the elimination of inorganic phosphate forming 5-enolpyruvylshikimate 3-phosphate (EPSP) catalyzed EPSP synthase (aroA-encoded EPSPS; EC 2.5.1.19). The seventh and final step in the main trunk of the shikimate pathway is the trans-1,4 elimination of phosphate from EPSP to yield chorismate catalyzed by chorismate synthase (CS; EC 4.6.1.4). The salicylate-derived mycobactin siderophores have been shown to be essential for M. tuberculosis growth in macrophages (de Voos et al. 2000). In addition, the shikimate pathway has more recently been shown to be essential for the viability of M. tuberculosis (Parish & Stoker 2002). Accordingly, the essentiality of mycobacterial shikimate pathway and its absence from the human host indicate that any of its enzyme components represents a promising target for the development of potentially non-toxic antimycobacterial agents. Incidentally, EPSP synthase enzyme is the site of action of glyphosate [N-(phosphonomethyl)glycine], which is a widely used-broad-spectrum herbicide (Steinrücken & Amrhein 1980). FINDING AND VALIDATING NEW DRUG TARGETS IN M. TUBERCULOSIS Although the complete genome sequencing of M. tuberculosis has been a landmark in mycobacterial research (Cole et al. 1998), the genetic determinants of M. tuberculosis virulence are not completely understood yet (Smith 2003). This is partly due to the difficulty in constructing mutants of M. tuberculosis, especially by gene replacement (Morsczeck 2003). However, the identification of essential genes, using either allelic exchange or transposon mutagenesis techniques, is of paramount importance, as it shows both the biological significance of the mutated gene and potential drug targets (Parish et al. 2001). The knowledge that a gene is essential for growth implies that the gene is an attractive molecular target, since its inhibition should kill the bacilli. The rearrangement of chorismate to prephenate, catalyzed by chorismate mutase (CM), is the first committed step in the biosynthesis of phenylalanine and tyrosine in bacteria, fungi, and plants (Dosselaere & Vanderleyden 2001). As this pathway is absent from mammals, it constitutes an attractive target for the development of new antimycobacterial agents. Although CMs are classified as `conserved hypothetical proteins' in the original genome annotation of M. tuberculosis, it has been proposed that there are probably two monofunctional chorismate mutases of the AroQ homology class in this pathogen (Calhoun et al. 2001). Chorismate mutases are generally intracellular metabolic enzymes required for the biosynthesis of phenylalanine and tyrosine, and the existence of an exported chorismate mutase in M. tuberculosis is puzzling. Homologues of exported chorismate mutases are generally present in parasitic or pathogenic organisms, suggesting that exported chorismate mutases may have evolved to participate in some aspect of parasitism or pathogenesis yet to be unraveled (Bekal et al. 2003). Accordingly, in order to both evaluate the role that may be played by these two enzymes and assess their use as potential targets for new antimycobacterial agent development, the aroQ (a probable cytoplasm-localized CM) and *aroQ (a probable periplasm-localized CM) coding sequences from M. tuberculosis H37Rv should be cloned and their biological activity determined. Single disruptions of either aroQ or *aroQ gene and double mutant aroQ/*aroQ should be generated to assess the relative importance of each chorismate mutase gene in the chorismate metabolism of M. tuberculosis. Homologues to enzymes in the purine salvage pathway have been identified in the genome sequence of M. tuberculosis (Cole et al. 1998). In the de novo synthesis of purine ribonucleotides, the formation of AMP and GMP from IMP is irreversible, but purine bases, nucleosides, and nucleotides can be interconverted through the activities of purine nucleoside phosphorylase (deoD, Rv3307), adenosine deaminase (add, Rv3313c), and hypoxanthine-guanine phosphoribosyl transferase (htp, Rv3624c). It has been suggested that inhibition of M. tuberculosis PNP could potentially lead to the accumulation of guanine nucleotides since a putative guanylate kinase (gmk, Rv1389) and nucleoside diphosphate kinase (ndkA, Rv2445c) are encoded in the genome (Basso et al. 2001). The synthesis and degradation of ppGpp [guanosine 3',5'-bis(diphosphate)], and pppGpp are catalyzed by (p)ppGpp synthase I (relA, Rv2583) using GTP as substrate (Avarbock et al. 1999). Increased concentration of hyperphosphorylated guanosine moieties is a central feature of a pleiotropic physiological response called the stringent response, through which bacteria enter a latent state in response to nutritional stress (Cashel et al. 1996). The accumulation of ppGpp has been implicated in the latency of M. smegmatis (Ojha et al. 2000). A rel-deficient mutant of M. tuberculosis was shown to no longer be capable of ppGpp synthesis and that this mutant had impaired long-term survival during in vitro starvation, indicating that ppGpp concentration may control mycobacterial adaptation to growth-limiting conditions, allowing for long-term survival (Primm et al. 2000). However, the role that deoD-encoded purine nucleoside phosphorylase may play in long-term survival (latency) of M. tuberculosis, if any, has not been established yet. The ability to regulate replication in host cells is a key element for many pathogens in establishing persistency. M. tuberculosis is a sophisticated intracellular pathogen, and different virulence factors related to entry and survival in macrophages are implied in the infection process. Polynucleotide phosphorylase (PNPase; EC 2.7.7.8) is an enzyme with polyadenylation activity that plays a key role in RNA maturation and degradation in E. coli and other bacteria. Recently, PNPase was found to affect mRNA levels of a set of virulence genes in Salmonella enterica, suggesting a possible connection between the enzyme activity and pathogenicity (Clements et al. 2002). A single point mutation in the PNPase coding sequence affected bacterial invasion and intracellular replication, and determine the alternation between acute or persistent infection in a mouse model for Salmonella enterica infection. We have identified an open reading frame in M. tuberculosis (gpsl. Rv2783c) that is homologous to bacterial PNPase gene sequences. However, there has been no report on the role of PNPase in M. tuberculosis. The bacterial heat-stable nucleoid-structuring (H-NS) protein is a small chromatin-associated protein that influences many cellular processes including recombination, transcription and transposition. H-NS has most extensively been studied in E. coli and it appears to exist to turn genes off (Dorman 2004). H-NS has affinity for all types of nucleic acids but binds preferentially to intrinsically curved DNA, which is commonly found at promoters, and can alter DNA topology by constraining negative supercoiling (Owen-Hughes et al. 1992). Mutations in hns - the gene that encodes H-NS - are highly pleiotropic. Many of the H-NS-modulated genes are related to adaptation to environmental conditions and/or virulence (Atlung & Ingmer 1997, Hommais et al. 2001). H-NS has been shown to be involved in repression of the virF virulence-gene promoter Shigella flexneri, a facultative intracellular pathogen (Falconi et al. 1998). Mutations in hns gene have been shown to be involved in high level expression of cholera toxin in Vibrio cholerae (Nye et al. 2000). More recently, H-NS has been shown to modulate expression of hemolysin, an important virulence factor of the opportunistic pathogen Serratia marcescens (Franzon & Santos 2004). In addition, deletion of the hns gene has been shown to confer drug resistance to E. coli by repressing multidrug exporter genes (Nishino & Yamaguchi 2004). The genome sequence of M. tuberculosis reveals a coding DNA sequence for H-NS (hns; Rv3852) (Cole et al. 1998). A study of the role played by H-NS in tuberculosis may unveil new drug targets involved in virulence. However, there has been no report yet on the role played by H-NS in TB. A diarylquinoline (R207910) has recently been shown to inhibit both drug-sensitive and drug-resistant M. tuberculosis in vitro with a minimum inhibitory concentration (MIC) value of 0.06 μg ml-1 , bactericidal activity in a murine TB model, and good safety and tolerability in humans (Andries et al. 2005). A puzzling feature of R207910, which represents a member of a new chemical class of antimycobacterial agents, is its exceptional specificity for its drug target: mycobacterial ATP synthase. M. tuberculosis and M. smegmatis strains resistant to R207910 were found to harbour a point mutation in the atpE gene, which codes for a part of the F0 subunit of ATP synthase. ATP synthase is a ubiquitous enzyme found in most living organisms, including humans. There is a very limited sequence similarity between the mycobacterial and human AtpE proteins, which bodes well for the satefy of the compound, as indicated by the phase I study in human volunteers (Andries et al. 2005). It has been pointed out that the direct application of the murine TB model to humans is still controversial because dormancy in the mouse essentially depends on the development of immunity, whereas in humans it is more likely due to low oxygen potential in lesions (Mitchison 2005). Nonetheless a new drug target has been unveiled for the development of new antimycobacterial agents and points to an ongoing need for validation of new targets to allow availability of a better arsenal of drugs to kill susceptible and resistant M. tuberculosis strains. Malaria continues to be a major cause of children's morbidity and mortality throughout the world, particularly in underdeveloped areas of Africa with up to three million deaths and approximately five billion episodes of clinical illness possibly meriting antimalarial therapy (Breman et al. 2004). More recently, an empirical approach to estimating the number of clinical events caused by P. falciparum worldwide, by using a combination of epidemiological, geographical and demographic data, has estimated that there were 515 (range 300-600) million episodes of clinical P. falciparum malaria in 2002 (Snow et al. 2005). These global estimates are up to 50% higher than those reported by the WHO. Malaria is caused by protozoan parasites of the genus Plasmodium (four species affecting humans) and transmitted by the Anopheles mosquito. P. falciparum is responsible for the most severe forms of malaria that frequently can be fatal if not conveniently treated. Malaria starts by inoculation of the Plasmodium parasite sporozoite stage that invade hepatocytes. Sporozoites transform into liver stages, and subsequent liver-stage development ultimately resulting in release of pathogenic merozoites that invade erythrocytes in the blood. In erythrocytes, the parasite undergoes cycles of asexual reproduction liberating new merozoites that invade and destroy new erythocytes. These repeated cycles of asexual reproduction of the parasite are responsible for the disease while the sequestration of mature forms of P. falciparum parasites inside the erythocytes at the endothelial cells of capilary vessels is responsible for severe malaria forms like cerebral and pulmonary malaria (Kappe et al. 2003). Treatment of P. falciparum malaria has depended for decades on the use of quinine. Synthetic antimalarial drugs, inspired by the knowledge of the quinine structure were developed in the 30ties and 40ties of the 20th century and permitted the development of the WHO world "erradication" program starting in the 50ties with large success in Europe and North America and many areas of Latin America, including malarial erradication in the coastal areas of Brazil. Two important groups of antimalarial drugs were particularly effective: the 4-aminoquinoline derivatives like chloroquine and amodiaquine and the synergistic association of antifolate (pyrimethamine) and sulphadoxine. Sulphadoxine is a sulphonamide that interferes with the action of dihydropteroate synthase (an enzyme in the folate biosynthesis pathway) and 2,4-diaminopyrimidine. Pyrimethamine is an inhibitor of dihydrofolate reductase enzyme activity. Malaria had been sheltered for many years from the dangers of resistance because of the outsdanding properties of chloroquine and the slow speed at which resistance developed to this drug (Wellems & Plowe 2001). However, resistance of P. falciparum to chloroquine, starting in the 60ties spread on a global scale and has exposed the ease with which resistance may develop to other drugs such as the antifolates (Sirawaraporn 1998). It has been pointed out that continued and sustainable improvements in antimalarial medicines through focused research and development are essential for the future ability to treat and control malaria worldwide (Ridley 2002). Unfortunately, malaria is a disease of poverty, and despite a wealth of scientific knowledge there is insufficient market incentive to generate the competitive, business-driven industrial antimalarial drug research and development that is normally needed to deliver new products. Mechanisms of partnering with industry should be established to overcome this obstacle and to open up and build on scientific opportunities for improved chemotherapy in the future. In the meantime, the best prospect for drug development and use in the short term is the government commitment to mechanisms and projects to ensure that improved drugs are sustainably discovered and developed and new targets for antimalarial agents are unveiled. Apicomplexan parasites are a large phylum of unicellular and obligate intracellular organisms of great medical importance. They include the human pathogens Plasmodium spp., the causative agent of malaria, and Toxoplasma gondii, an opportunistic parasite of immunosuppressed individuals and a common cause of congenital disease. Apicomplexan parasites also include parasites of veterinary importance that cause considerable economic losses such as Babesia bovis, Babesia bigemina, Theileria parva, and Theileria annulata, which are pathogens of domestic livestock, and coccidial parasites belonging to the genus Eimeria, with seven species, a major cause of acute disease and ill thrift in poultry, cattle and sheep (Shaw 2003, Shirley et al. 2004). Completion of the genome sequence of P. falciparum, and growing sequence databases for other Plasmodium species, T. gondii, E. tenella, T. parva, and T. annulata should provide valuable tools for whole-genome analysis of Apicomplexa to allow identification of new targets for drug development (Gornick 2003). The search for new and effective drugs against these pathogens has been boosted during the last years by an unexpected finding. Through molecular and cell biological analysis it was realized that probably most members of this phylum harbor a plastid-like organelle, called the apicoplast, which probably is derived from the engulfment of a red algae in ancient times. Although the apicoplast itself contains a small circular genome, most of the proteome of this organelle is encoded in the nuclear genome, and the proteins are subsequently transported to the apicoplast. It is assumed to contain a number of unique metabolic pathways not found in the vertebrate host, making it an ideal source of drug targets (reviewed by Seeber 2003). Amongst them are three enzymes of the plant-like fatty acid synthesis machinery (acetyl-CoA carboxylase, β-ketoacyl-ACP synthase and enoyl-ACP reductase), suggesting that fatty acid and lipid biosynthesis is a major function of the apicoplast. Moreover, completion of the P. falciparum genome sequence (Gardner et al. 2002) and availability of a Plasmodium genome database (http://www.PlasmoDB.org) (Bahl et al. 2002) should provide valuable tools for discovery of new drug targets. More specifically, avian coccidia constitute a problem with serious economic and social consequences in view of the dramatic epidemic nature of the disease that is responsible for high mortality in chicken breeding colonies. According to Martin Shirley (Shirley et al. 2004) from the Institute of Animal Health (Compton, UK), approximately 35 x 109 chickens are consumed every year in the world and each one harbors approximately 5 x 106 Eimeria which gives 175 x 1015 parasites, making Eimeria one of the most numerous organisms in the planet. More recently, the interest in coccidia has shifted to Cryptosporidium and Toxoplasma but Eimeria continues to represent a major veterinary concern due to economic losses resulting from this pathogen. Genome sequencing projects of E. tenella in the Sanger Institute (UK) and in Texas A&M University (Veterinary, Pathobiology, US) have identified some molecular targets such as manitol 1-phosphate dehydrogenase from the manitol cycle that seems to play an important role in the sexual cycle of the parasite, which thus may represent a target for parasite maturation and transmission. Another interesting target is a histone deacetylase that is the molecular target of the antibiotic apicidin, which is active against plasmodial parasites and also against Eimeria. These proteins may represent molecular targets for immobilization on solid support in natural-product screening efforts. VALIDATED
TRAGETS FOR ANTIMALARIAL AGENTS The rationale for targeting enzyme components of the type II fatty acid synthase system (FAS-II) was given above. Here we focus on one FAS-II enzyme component: enoyl-ACP reductase. Triclosan, a chlorinated bisphenol that is widely used in many contemporary consumer and professional health care products, has been reported to inhibit the in vitro growth of P. falciparum with an IC50 value of 150-2000 ng ml-1 (McLeod et al. 2001). Triclosan has also been shown to offer protection against blood stages of malaria and that the drug target is an enoyl-ACP reductase from P. falciparum (Surolia & Surolia 2001). This enoyl reductase was shown to be NADPH-dependent enzyme and accepts acetoacetyl-CoA as co-substrate (Pillai et al. 2003). More recently, triclosan has been shown to act as a slow-tight binding inhibitor of the P. falciparum enoyl-ACP reductase enzyme activity with an overall inhibition constant value of 96 pM (Kapoor et al. 2004). Determination of the three-dimensional structure of malarial enoyl reductase-triclosan NAD+ ternary complex has provided a structural framework that sheds light on the mode of binding of triclosan (Perozzo et al. 2002). Accordingly, P. falciparum enoyl-ACP reductase is a validated target for antimalarial agent development and to be used in drug screening efforts. THE SHIKIMATE PATHWAY The discovery of a functional shikimate pathway in aplicomplexan parasites, P. falciparum and T. gondii, provides several targets for the development of new anti-parasite agents (Roberts et al. 1998). In vitro inhibition of the growth of P. falciparum by the herbicide glyphosate was reversed by treatment with p-aminobenzoate, suggesting that the shikimate pathway supplies folate precursor for P. falciparum growth. Two shikimic acid analogs, 6-S-fluoroshikimate and 6-R-fluoroshikimate, have been shown to inhibit P. falciparum growth and inhibition shown to be specific to the shikimate pathway (McConkey 1999). Despite detection of enzymatic activities of six out of seven shikimate pathway enzymes in crude extracts of P. falciparum and T. gondii (Dieckman & Jung 1986, Roberts et al. 1998), only a single gene encoding the terminal enzyme in the shikimate pathway has been identified in the genome annotation. The coding DNA sequence of chorismate synthase has been identified in P. falciparum and shown to be located in the parasite cytosol (Fitzpatrick et al. 2001). P. falciparum chorismate synthase is "monofunctional", requiring an external source of reduced flavin mononucleotide, similar to plants and bacteria. Chorismate synthase has been shown to be required for normal growth, and disruption of expression by RNA interference decreases parasite growth (McRobert et al. 2002). Hence, P. falciparum chorismate synthase is a validated target for drug development and amenable to large-scale production for drug screening efforts. The existence of a bifunctional EPSPS-SK enzyme in Plasmodium species has recently been suggested (McConkey et al. 2003). However, evidence for the biological activity of the gene product still awaits experimental proof. PURINE SALVAGE PATHWAY An essential step in the life cycle of protozoan parasites is cellular replication of the trophozoite in the human erythrocyte - asexual blood stage of P. falciparum - that creates a need for large quantities of purines for RNA and DNA synthesis. However, P. falciparum lacks de novo purine synthesis (purine auxotroph), and starvation of purines is known to cause purine-less death in cultured cells (Reyes et al. 1982, Kicska et al. 2002a). Enzymes of the purine salvage pathway were detected in P. falciparum including purine nucleoside phosphorylase (PNP), and thus the parasite must salvage purine bases from the mammalian host to survive. The demand for purine bases makes growth of Plasmodium sensitive to disruption of pathways for purine salvage (Wiesmann et al. 1984). Hypoxanthine has been reported to be the major purine precursor for purine salvage, and P. falciparum growth is reduced by culturing in the presence of xanthine oxidase, which depletes both the medium and erythrocyte of hypoxanthine (Berman et al. 1991). The sole pathway of hypoxanthine production in P. falciparum and in human erythrocytes is through the phosphorolysis of inosine to hypoxanthine, a reaction catalyzed by PNP (Sherman 1979). Immucillin-H, a transition state analogue that inhibits PNP enzyme activity, has been shown to both target P. falciparum PNP reducing the incorporation of inosine into nucleic acids and kill P. falciparum cultured in human erythrocytes (Kicska et al. 2002a). Immucillin-H has been shown to be a slow-onset tight binding inhibitor of recombinant P. falciparum PNP (Kicska et al. 2002b). More recently, determination of the crystal structure of P. falciparum PNP in complex with immucillin-H and sulphate ion reveals that its active site presents structural differences from human PNP (Shi et al. 2004). An inhibitor, 5'-methylthio-immucillin-H, more specific for P. falciparum PNP as compared to human PNP has been described (Shi et al. 2004). It has more recently been demonstrated that 5'-methylthio-immucillin-H kills P. falciparum strain 3D7 cultures and that inhibition of parasite growth occurs at concentrations where P. falciparum PNP but not human PNP is strongly inhibited (Ting et al. 2004). Interestingly, it has been proposed that the parasite PNP enzyme may lie at an essential metabolic branchpoint, generating hypoxanthine from both the purine nucleoside pathways and from polyamine synthesis based on its unusual substrate specificity (Shi et al. 2004, Ting et al. 2004). These results warrant structure-based design studies and screening efforts aiming at discovering and developing malaria-specific compounds targeting the parasite PNP enzyme. OTHER POTENTIAL TARGETS FOR NEW ANTIMALARIALS A number of molecular targets can be related to the functions of distinct organelle structures of Plasmodium spp. asexual blood stage (reviewed by Bannister et al. 2000). The lysosomal food vacuole (the site of extensive haemoglobin degradation), the apicoplast (a plastid organelle thought to originate from a green algal symbiont), and an acrystate mitochondrion with a limited electron transport system harbour molecular targets of interest for drug development. For instance: plasmepsin aspartic proteases, cysteine protease falcipain 2 and metallopeptidase falcilysin in the vacuole; non-mevalonate isoprenyl biosynthesis (link to protein prenylation) and plastid DNA replication and transcription in the apicoplast; and dihydroorotate dehydrogenase in the mitochondrion. In addition, the current understanding of mutations in dihydrofolate reductase that confer resistance to pyrimethamine/sulphadoxine, the ability to model the dihydrofolate reductase active site, and the ease of dihydrofolate reductase enzyme assay suggest that further improved inhibitors are achievable (Ridley 2002). Functional thioredoxin and glutathione systems have been shown to participate in antioxidant defence in P. falciparum, thereby representing an interesting drug target because of the necessity of the parasite to deal with the large amount of haem released by hemoglobin degradation in the digestive vacuole. Glutathione S-transferases (GST) catalyze the conjugation of glutathione with a wide variety of hydrophobic compounds, generally resulting in non-toxic products that can be readily eliminated. In contrast to many other organisms, P. falciparum possesses only one GST isoenzyme. Plasmodial GST is a promising target for antimalarial drug development because it is highly abundant in the parasite, its activity was found to be increased in chloroquine-resistant cells, and it has been shown to act as a ligand for parasitotoxic hemin. The crystal structure of P. falciparum GST has been solved at 1.9 Å resolution and shown to differ considerably from the human enzyme (Fritz-Wolf et al. 2003), a feature that may be exploited to search for specific inhibitors. Studies on the activity of ferriprotoporphyrin IX (FP) on redox-active enzymes (Campanale et al. 2003) indicates that it inhibits glycolysis and activates the pentose phosphate pathway, which is the only source of NADH for the parasite. P. falciparum lactate dehydrogenase (LDH) is essential for NAD+ regeneration and therefore represents a potential drug target to be used in screening of extracts and plant-derived natural products. Iron chelators have been shown to suppress plasmodial growth in culture (Hershko et al. 1991). Desferrioxamine B (DFO B) has been used in clinical trials for the control of P. falciparum malaria infection with partial positive results (Gordeuk et al. 1992). These results prompted an intensive study of synthetic siderophores of the family of mono catecholates (dihydroxybenzoates), aminoacids, hydrazones of the glyoxylic acid, phenylglyoxylic or formyl benzoic acids (Rotheneder et al. 2002). LD50 values ranging from 10 nM to 100 mM were obtained; however, high toxicity was observed for K562 mammalian cells. Reversibility of anti-plasmodial activity was observed for some, but not all, siderophores in the presence of iron salts. A study on dicatecholate FR160 anti-plasmodial activity showed IC50 values ranging from 0.8 to 1.5 µM (Pradines et al. 2002). Interestingly, these auhors showed that increased liposolubility does not lead to improved anti-malarial action. Ferriprotoporphyrin (FP) derived from haemoglobin is rapidly polymerized to hemozoin, but it has been demonstrated that only 55% of FP was polymerized and that the non-polymerized FP leaving the digestive vacuole is degraded by glutathione and is the source of iron for the parasite (Ginsburg 1999). It has been pointed out that there is no doubt that the parasite acquires iron from intracellular sources (Ginsburg et al. 1998, Ginsburg & Golenser 2003). However, as the total amount of iron in the parasite remains unchanged during parasite growth, it is necessary to define the mechanism of iron sequestration by the parasite. In summary, it can be said that different siderophores appear to have different mechanisms of action at different levels of the iron uptake process and that natural siderophores (such as pioverdine of Serratia and catecholamines of Klebsiella) seem to have particular mechanisms not entirely reproduced by synthetic siderophores. In the screening of natural compounds against P. falciparum here proposed, these features will be taken into consideration and natural siderophores will be tested in culture media without human serum, with and without iron chelators (ferritin, dipiridyl), and with or without iron salts (FeCl3 and FeSO4). T-CELL IMMUNOMODULATION Mutations in the locus encoding for purine nucleoside phosphorylase (PNP) cause gradual decrease in T-cell immunity, though keeping B-cell immunity normal as well as other tissues (Stoop et al. 1977). The activation of helper T cells requires that they recognize a complex formed between an antigen and a class II MHC (Major Histocompatibility Complex) protein on the surface of antigen-presenting cells with appropriate costimulation. This results in interleukin-2 release which, in turn, leads to T-cell clonal expansion with activity against cells exhibiting the stimulatory antigen (Levinson & Jawetz, 2000). However, most naïve T-cells receive no antigenic signal and undergo apoptosis. Cellular nucleic acids from the apoptosed cells are recycled. Naïve T-cells have the ability to transport and phosphorylate deoxyguanosine (dGuo) to deoxyguanosine triphosphate (dGTP), which accumulates relative to normal cells (Bantia et al. 1996). Two enzymes that utilize dGuo as a substrate in humans are PNP and deoxycytidine kinase (dCyK). PNP catalyzes the phosphorolysis of dGuo to guanine and ribose 1-phosphate. The normal role of dCyK in dividing T-cells is the salvage of deoxycytidine to form deoxycytidine triphosphate (dCTP). dGuo predominantly undergoes phosphorolysis by PNP because dGuo has a higher affinity for PNP than dCyK (Krenitsky et al. 1976). When dGuo accumulates beyond normal levels, dCyK catalyzes the conversion of dGuo to deoxyguanosine monophosphate (dGMP), which is then converted to dGTP. The allosteric inhibition site for dGTP on ribonucleotide diphosphate reductase inhibits cellular formation of dCDP and dUDP, thereby preventing DNA synthesis. Inhibition of PNP enzyme activity leads to an increase in dGuo concentration that will, in turn, be converted to dGTP by dCyK enzyme activity and ensuing accumulation of dGTP in T-cells. dGTP accumulates within T-cells because of the inability of nucleotides to cross the cell membrane (Bantia et al. 1996). Accumulated dGTP inhibits ribonucleotide reductase, thus preventing the conversion of ribonucleoside diphosphates to corresponding deoxyribonucloside diphosphates (Fig. 6). Depletion of deoxyribonucleotides ultimately results in the inhibition of DNA synthesis and cell replication resulting in suppression of proliferation of immature T-cells (Kazmers et al. 1981). In summary, the absence of PNP activity leads to impaired cellular proliferation required for an immune response (Mitchell et al. 1978). Type IV autoimmune disorders are a primary disease target for PNP inhibitors, and are caused by inappropriate activation of T-cells by self-antigens (Schramm 2002). These disorders include rheumatoid arthritis, psoriasis, inflammatory bowel disorders and multiple sclerosis. In addition, T-cell proliferative disorders, including organ transplant rejection, cutaneous T-cell lymphoma, and adult T-cell leukemia, would be primary proliferative targets for PNP inhibitors (Bantia et al. 1996). Accordingly, a transition-state analog (immucillin-H; trade name BCX-1777) that inhibits human PNP enzyme activity has recently been shown to inhibit the growth of malignant T-cell leukemia cell lines with the induction of apoptosis in the presence of dGuo (Kicska et al. 2001). More recently, we have determined the three-dimensional structure of human PNP in complex with immucillin-H which should allow a structural basis for the design of more potent PNP inhibitors (Azevedo et al. 2003b). In addition, some PNP inhibitors have been tested in combination with nucleoside antiviral and anticancer drugs showing the ability to potentiate the in vivo activity of these drugs (Bennett et al. 1993). Purine nucleoside phosphorylase (PNP; EC 2.4.2.1) is a key enzyme of the purine salvage pathway (Fig. 7), responsible for the inter-conversion between (deoxy) nucleosides and bases, which in turn may be converted to uric acid for excretion or reused in nucleic acid biosynthesis (Parks & Agarwal 1972). PNP catalyzes the reversible cleavage, in the presence of inorganic phosphate (Pi), of N-ribosidic bonds of purine nucleosides and deoxynucleo-sides, except adenosine, to generate ribose 1-phosphate and the corresponding purine base (Kalckar 1947). Human PNP is inactive against pyrimidine (deoxy) nucleosides. The major physiological substrates for mammalian PNP are the natural 6-oxypurines inosine, guanosine, and 2'-deoxyguanosine (Schramm 1998). Human PNP is specific for purines nucleosides in the β-configuration and exhibits a preference for ribosyl-containing nucleosides relative to the analogs containing the arabinose, xylose, and lyxose stereoisomers (Stoeckler et al. 1980). Moreover, cleavage of glycosidic bond proceeds with inversion of configuration, from β-nucleosides to a-ribose 1-phosphate (Fig. 8; Porter 1992). Thermodynamically, the equilibrium of the reaction is shifted in favour of nucleoside synthesis. However, in vivo phosphorolysis is highly favoured over synthesis, owing to coupling with oxidation of purine bases catalyzed by xanthine oxidase (XOX) and phosphoribosylation catalyzed by hypoxanthine-guanine phosphoribosyl transferase (HGPRT) (Fig. 7). The catalytic efficiency of mammalian PNPs is high for the deoxyguanosine nucleoside that characterizes its biological function. The homotrimer exhibits Michaelis-Menten initial rate kinetics and has no known physiologic regulatory sites. PNP is present at micromolar concentrations in blood cells and is coupled to a substrate trapping phenomenon known as catalytic commitment (Kline & Schramm 1993). Every collision of a substrate (deoxy) nucleoside with the catalytic site leads to its trapping and conversion to product. With the high concentrations of enzyme, catalytic commitment, and low Michaelis-Menten constant (Km) value, cells containing high concentrations of PNP are assured of the virtual absence of free deoxy-guanosine. Mitochondrial deoxyguanosine metabolism is exempt from this degree of deoxyguanosine removal since PNP is absent and a deoxyguanosine kinase is present (Arpaia et al. 2000). Repair and recycling of mitochondrial DNA generates deoxyguanosine that is proposed to remain in this compartment (Schramm 2002). As pointed out above, Immucillin-H (Fig. 8), a potent PNP inhibitor based on transition-state structure of enzyme-catalyzed chemical reaction, inhibits the growth of malignant T-cell leukemia cell lines (Kicska et al. 2001). The chemical structure of Immucillin is based on steady-state and pre-steady state results as well as on kinetic isotope effects (reviewed in Schramm 2005). The results established that PNP stabilized a ribooxocarbenium ion and protonated N7 of the purine base to establish the transition state. The features of both guanine and hypoxanthine were incorporated into the design of Immucillin-H, but with elevated pKa value at the N7 position accomplished by the use of 9-deazapurines (Fig. 8). The charge or H-bond potential of the ribooxocarbenium ion is provided by the iminoribitol moiety with a stable ribosidic bond (Fig. 8). Immucillin-H is a slow-onset, slow-release, tight-binding inhibitor for mammalian purine nucleoside phosphorylase with picomolar dissociation constants (Miles et al. 1998). Human PNP is classified, based on substrate specificity and structural characteristics, into the Nucleoside Phosphorylase-I family, which includes nucleoside phosphorylases that share a common α/β-subunit fold and show either a trimeric or a hexameric quaternary structure (Pugmire & Ealick 2002). The crystallographic structure of human PNP was first determined in 1990 at 3.2 Å resolution (Ealick et al. 1990). Further crystallographic studies improved the resolution to 2.8 Å (Ealick et al. 1991). These atomic coordinates were extensively used for structure-based design of PNP inhibitors (Woo et al. 1992, Chern et al. 1993, Guida et al. 1994, Morris et al. 2000). However, the deposited atomic coordinates of human PNP (PDB access codes: 1ULA and 1ULB) were recently withdrawn due to low resolution. We have recently cloned, overexpressed and purified to homogeneity functional human PNP (Silva et al. 2003). Collection of X-ray diffraction patterns allowed determination of the crystal structure of the recombinant human PNP using synchrotron radiation and cryocrystallographic techniques at higher resolution than previously reported (Azevedo et al. 2003a). This structure provided a more precise picture of the enzyme active site and represents a more reliable picture on which to base structure-based design of PNP inhibitors. Mathematical predictions suggest that inhibitors based on the transition-state structure for PNP will bind with dissociation constant values in the attomolar (10-18 M) range (Miles et al. 1998). We have also determined the three-dimensional structure of human PNP in complex with immucillin-H (a picomolar inhibitor) which should allow a structural basis for the design of even more potent PNP inhibitors (Azevedo et al. 2003b) SCREENING OF NATURAL-PRODUCT COMPOUNDS The analysis of molecular interactions is a key event in the development of new pharmaceutical and biotechnological products as well as in exploratory life science research. Accordingly, millions of dollars are spent early in drug development on screening compounds for receptor binding in vitro. Biosensors are commonly used for such tasks and can give detailed information on the binding affinity and in many cases also on the binding kinetics of an interaction between chemical compounds and their targets. At present, most screens that are used in drug discovery require some type of fluorescent labelling or radiolabelling to report the binding of a ligand to its receptor. This labeling step has several disadvantages, including extra time, cost demands and some interference in assays. Ideally, a biosensor-based screening platform should be label-free, sensitive and have sufficient throughput to be widely applicable in drug discovery (Cooper 2002). The real-time BIA from Pharmacia Biosensor AB (BIACORE), which is a label-free technology for monitoring biomolecular interactions as they occur, was chosen for high-throughput screening of natural-product compounds that interact with the validated drug targets described above. The detection principle of BIACORE equipment relies on surface plasmon resonance (SPR), an optical phenomenon that arises when light illuminates thin conducting films under specific conditions. The use of Surface Plasmon Resonance (SPR) technology, which is an optical label-free detection technique, makes it possible to observe the binding of an analyte molecule to a given surface-immobilized target molecule. Typical target molecules are soluble receptors, binding proteins, enzymes, peptides, and DNA (Karlsson et al. 2000). Since the introduction of the BiacoreTM 'biosensor' system in the early 1990s, SPR and related techniques have gained tremendous popularity. The BIACORE's SPR technology has been designed to investigate the functional nature of binding events. The reliability and success of BIACORE is built on three cornerstone technologies: sensor chip technology, microfluidics, and SPR detection system. Quantitative measurements on the binding interaction between two or more molecules are dependent on the immobilization of a target molecule to the sensor chip surface. Binding partners to the target can be captured from a complex mixture, in most cases, without prior purification as they pass over the chip. The sensor chip consists of a glass surface coated with a thin layer of gold. In the most widely used sensor chip, the gold surface is modified with a carboxymethylated dextran layer. This dextran hydrogel layer forms a hydrophilic environment for attached biomolecules, preserving them in a non-denatured state. A range of other derivatized surfaces is also commercially available to enable various immobilization chemistries. BIACORE sensor chips provide reproducible results, high chemical stability and low non-specific binding. The chemical stability allows regeneration of sensor chip surfaces for many cycles depending on the nature of immobilized ligands (100 is average), and they can withstand high salt concentrations, extremes of pH and organic solvents. The Integrated micro Fluidics Cartridges (IFC) allow analyte (e.g., plant extracts and/or isolated natural products) to pass over the sensor surface in a continuous, pulse-free and controlled flow, thereby maintaining constant analyte concentration at the sensor chip surface. IFC allows low sample consumption, absence of an air-solution interface where samples can evaporate and proteins can be denatured, free analyte concentration is constant and therefore known at all times, no washing steps are needed to replace the sample with buffer, a range of surface ligand concentrations and contact times can be analysed in one experiment improving kinetic and concentration analysis, and accurate temperature stability, which is of paramount importance to obtain meaningful innibition constants. The gold layer in the sensor chip creates the physical conditions required for SPR. Essentially, SPR detects changes in mass in the aqueous layer close to the sensor chip surface by measuring changes in refractive index. When molecules in the test solution bind to a target the molecular mass increases, when they dissociate the mass falls, and this simple principle forms the basis of the sensorgram - a continuous, real-time monitoring of the association and dissociation of the interacting molecules. The sensorgram provides quantitative information in real-time on specificity of binding, active concentration of molecule in a sample, kinetics and affinity. Binding kinetics with rate constants ranging from 1 s-1 to 1x10-4 s-1 can be studied, and equilibrium dissociation constants between 100 pM and 100 µM can be quantified. A wide range of molecules can be analyzed, from low-molecular-mass drugs to multiprotein complexes and bacteriophage (Cooper 2002). Molecules as small as 100 Daltons can be studied. Importantly, as previously pointed out, there is no need to label molecules with fluorescent or radioactive tags, so avoiding the possibility that labels may compromise activity. Molecules can be studied in their native state to provide results that reflect in vivo activity. Moreover, biosensor technology has other advantages including low sample consumption, simplified sample handling, and relatively rapid and automated data analysis (Frostell-Karlsson et al. 2000, Cooper 2002). Thus, solid-phase detection techniques potentially cope with today's need for multiplexing and automatizing binding assays for high-throughput analysis (Piehler 2005). BIACORE's SPR technology within the scope of the present study can be used to identify binding partners to any target molecule (ligand fishing) and "hit" to lead characterization (rapid affinity ranking and detailed kinetics of interaction for small molecules binding to target proteins). The BIACORE 3000 Control software includes a microrecovery function designed for recovering material from the sensor surface in a very small volume (3-7 μl) to be analysed by mass spectrometry, which facilitates identification of ligand specifically bound to immobilized proteins from cell and tissue extracts (Piehler 2005). There are three main types of coupling chemistry which utilize amine (e.g., lysine), thiol (cysteine) or aldehyde (carbohydrate) functional groups. All covalent coupling methods utilize free carboxymethyl groups on the sensor chip surface. They can therefore be used for any of the sensor chips that have such carboxymethyl groups (e.g., Sensor Chip CM5). The side chain of lysine is a hydrophobic chain of four methylene groups capped by an amino group that ionizes with an intrinsic pKa value of 11.1 in the absence of perturbing factors, so it is ionized under most physiological conditions. There is always a finite fraction of nonionized amino groups, however, which are potent nucleophiles. Consequently, the amino groups of lysine residues readily undergo a variety of acylation, alkylation, arylation, and amidination reactions. Acetylation of amino groups is possible with a variety of anhydrides - acetic, succinic, maleic, citraconic, and 3,4,5,6-tetrahydrophthaloyl. Direct immobilization of protein ligands is possible through linkages between the N-hydroxy-succinimide (NHS) ester groups on a hydrophilic dextran matrix of BIACORE and amine groups on proteins. In protein molecules, NHS ester cross-linking reagents couple principally with the a-amines at the N-terminals and the e-amines of lysine side chains (Hermanson 1996). For instance, human PNP protein has 12 lysine residues in its primary sequence, it is likely that immobilization of the recombinant protein will not present difficulties. If the protein to be immobilized has a surface-exposed disulphide or a free cysteine, ligand-thiol coupling is the method of choice. The thiol group of cysteine residues is the most reactive of any amino acid side chain. The cysteine thiol usually ionizes at slightly alkaline pH values with an intrinsic pKa in the region of 9.0 to 9.5. The carboxymethyl groups on the sensor surface are activated by NHS, thus creating a reactive succinimide ester that reacts with 2-(2-pyridinyldithio)ethane-amine (PDEA) to introduce a reactive disulphide group, which, in turn, reacts with free cysteine on the protein to be immobilized. Only a modest amount (5-10 mg) of protein is needed for covalent immobilization on the sensor chip. The major requirement is that the protein is pure and has a high level of activity because direct coupling is relatively indiscriminate and protein contaminants will also be coupled. Another requirement is that protein stocks should be fairly concentrated (> 0.5 mg ml-1). These requirements are met by a number of reports on recombinant proteins we have published (Basso et al. 1998, 2001, Azevedo et al. 2003a,b, Oliveira et al. 2003,2004, 2005, Santos et al. 2003, Silva et al. 2003, Dias et al. 2004, Nolasco et al. 2004, Pereira et al. 2004a,b, Rizzi et al. 2005). CRYSTALLOGRAPHIC SCREENING In order to identify possible new lead compounds against diseases such as malaria and tuberculosis, we will use the crystallographic screening procedure (Nienaber et al. 2000). The determination of the structural basis for specificity of ligands against protein targets is a scientific endeavor that makes use of computational (Kuntz et al. 1994) and experimental approaches (Nienaber et al. 2000). The interaction between protein and ligand is the key feature in the structure-based design of inhibitors. However, the interaction between protein and ligand can only be structurally defined applying crystallography and NMR spectroscopy. Furthermore, the precise definition of the ligand binding demands structural study of several complexes between protein and different ligands, which may demand several years of structural studies (Canduri & Azevedo 2005). Methods allowing high-throughput screening could be applied to speed up the process of determination of structural basis for specificity of ligand library for a selected protein target. The method is rapid, efficient, and high-throughput, and it results in detailed crystallographic structure information (Nienaber et al. 2000). Crystallography screening opened the possibility of testing several potential ligands for a protein target, which accelerates the identification of new ligands. The process demands a great amount of purified protein and has been used to identify new inhibitors for urokinase (Reuning et al. 1998). This method has been used to sample large compound libraries and detects ligands by monitoring variations in the electron density map relative to the native form. Nevertheless, even using high concentrations of ligand there are several examples of failure of obtaining complex between a protein target and a high-affinity ligand (Nienaber et al. 2000). The main reasons for this failure are the following: (1) low solubility of the ligand, (2) large conformational changes due to ligand binding, which may break crystals (in the case of soaking experiments), and (3) small solvent channels in the crystal or obstructed binding site. Therefore, a protein target must satisfy several conditions to be used in the crystallographic screening of ligands, mainly crystals with high-solvent content and high-resolution diffracting crystals. An interesting application of crystallographic screening is the study of the structural basis for binding of identified ligands. In this application the step of exposing the crystal to a mixture of compounds may be omitted. We have recently applied crystallographic screening in order to identify the structural basis for differences in the specificity of several ligands of human PNP. Crystals of human PNP are suitable for crystallographic screening of ligands, since they show large solvent channels (solvent content up to 75%), which allows diffusion of ligands in the crystal lattice, very efficient protein expression making protein availability not a limiting step, and high-resolution diffraction crystals. Fig. 9 describes the overall scheme used to study the structural basis specificity of ligands for human PNP. To increase the sampling rate of the crystallographic screening method, compounds are tested as specially designed mixtures. These mixtures are designed to capitalize on the high-resolution structural data available from X-ray crystallography. Specifically, the raw experimental data (electron density map) provides the shape of the bound compound and can therefore provide the identity of the ligand in a mixture if the mixture is suitably designed to be shape-diverse such as those found in plant extracts. Recombinant proteins can be expressed and purified. The proteins to be studied have to be crystallized using vapour diffusion and crystallization conditions as previously described (Azevedo et al. 2003a,b,c, Santos et al. 2003, Canduri et al. 2004, 2005, Dias et al. 2004, Nolasco et al. 2004, Pereira et al. 2004a,b). Usually, the solution containing ligands is dissolved and the native crystals are soaked in these solutions 48 h prior to data collection. The ligand which shows the highest specificity for the crystallized protein will bind to the active site. The next steps are focused on the solution and analysis of the binary complex structure. In order to increase the resolution of the crystals,the data is collected from a flash-cooled crystal at 104 K. Prior to flash cooling, glycerol is added, up to 50% by volume, to the crystallization drop. X-ray diffraction data is collected at a wavelength of 1.4310 Å using the Synchrotron Radiation Source (Station PCr, Laboratório Nacional de Luz Síncrotron, LNLS, Campinas, Brazil) and a CCD detector (MARCCD). X-ray diffraction data is processed up to highest resolution using the program MOSFLM and scaled with the program SCALA (CCP4 1994). The standard procedure of molecular replacement using AMoRe (Navaza 1994) is currently used to solve protein structures, when necessary. Further refinement in CNS (Brunger et al. 1998) will continue with simulated annealing using the slow-cooling protocol, followed by alternate cycles of positional refinement and manual rebuilding using XtalView (McRee 1999). Finally, the positions of water and ligand molecules are checked and corrected in Fobs - Fcalc maps. Analysis of the structure enables identification of the bound ligand, and the structure may provide new insights to improve inhibitor specificity. CHEMICAL DERIVATIZATION OF NATURAL COMPOUNDS AND SAR STUDIES The challenge of
any drug discovery effort is to identify and develop compounds with properties
that are predictive of good efficacy and safety in humans. In this regard,
organic synthesis plays a pivotal role. Once lead series with some desirable
profiles are identified, the compounds can progress to lead optimization,
entailing structural modifications with the goal of achieving optimal efficacy
and pharmacokinetic/pharmacodynamic properties. The frequent occurrence of
natural products as complexes of structurally related analogues can be exploited
by the natural-product investigator as a guide for initial SAR (Structure-Activity
Relationship) experiments. In this regard, even simple synthetic modifications,
as those obtained through "shotgun" transformations (key functional
groups required for antibiotic activity can be identified by allowing the
parent compound to react with nonspecific derivatizing reagents, such as
alkyl halides, anhydrides, acyl halides) can be instrumental in leading to
an optimized semi-synthetic analogue. The knowledge gained through understanding
the natural SAR and the shotgun approach can provide an early foundation
on which an overall synthetic strategy could be developed. An illustration
of this principle is found in the mannopeptimycin family of antibiotics (Fig.
10), a complex of glycopeptides produced by the Streptomyces hygroscopicus strain
LL-AC98 that shows activity against methicillin-resistant Unlike simpler synthetic compounds, natural products can be limited in supply owing to source limitations or the impracticality of total synthesis. Some plant-derived agents are amenable to culturing on a production scale, and synthetic methodologies continue to be developed for the large-scale synthesis of highly complex products. One of the most compelling recent examples is that of discodermolide, an antitumor polyketide from the Caribean sponge Discodermia dissoluta (Gunasekera et al. 1990) (Fig. 11). The low isolation yield of (+)-discodermolide (~14 mg/ kg), combined with the limited natural supply of the producing organism, made it necessary to pursue chemical synthesis as a means to provide the compound for further development. Following the initial report of its isolation, the compound quickly became a target for several academic groups, and several successful syntheses were reported. Finally, by blending several different methodological approaches (Marshal & Johns 1998, Paterson et al. 2000, Smith et al. 2000) the team at Novartis succeeded in synthesizing 60 g of (+)-discodermolide. Successes such as these make the compelling case that virtually no crucially important compound is beyond reach for clinical evaluation. In the course of both shotgun experiments or total synthesis it is often possible to define the crucial structural elements required for biological activity. In this sense, potent and selective products can be derived with fewer synthetic steps and at a reasonable cost. In some cases, a natural product has been shown to have high potency for a particular target, but is not practical for use as a therapeutic agent, owing to various liabilities, for example, supply issues, pharmacokinetic/phamacodynamic liabilities and so on. In these cases, understanding the binding interactions of the natural product and the target can lead to a model for synthetic mimetics. An example of this approach to mimetics of the cryptophycin antitumor agents started with an azepine scaffold to which residues were attached that resembled the overall geometry in the natural product (Smith et al. 2003). A synthetic strategy was developed that allowed the azepine mimetic compound to be prepared and with a reasonable overall yield. In this process, the stereochemical arrangement of the side chains and side-chain composition were studied to optimize the biological response. The increasing demand of new drugs translates directly into a need for new methods to rapidly assemble highly pure molecules that possess an ever-increasing level of structural complexity. These processes are also required to be environmentally cleaner (Green Chemistry), more efficient and lead to greater structural variation in as short a period of time as possible. Such demands have driven the development of novel technologies, which have begun to produce compounds at a greater rate than previously thought possible. One such molecule-assembly technology is that of solid-supported reagents. The use of solid supported reagents in chemical synthesis in a multistep mode has been shown to markedly improve productivity in crucial aspects of the generation of fine chemicals and complex target molecules (Ley & Baxendale 2002). In this regard, instead of viewing natural products as a stand-alone approach distinct from combinatorial synthesis, it is now much more effective to adopt strategies that combine both approaches. In principle, there seem to be a number of strategies through which the unique molecular diversity of natural products can be leveraged in the design of combinatorial libraries. The target-orientated or focused-library approach seeks to elaborate structural modifications onto an existing bioactive natural product scaffold in a parallel, systematic fashion, in order to improve its inherent biological activity or drug-like properties. This can be performed either by semi-synthetic modification of the parent molecule, or by fully synthetic methods (Kissau et al. 2003). The diversity-oriented approach seeks to leverage the privileged structural motifs of natural product scaffolds to synthesize combinatorial libraries capable of binding a wide range of biological targets (Burke & Schreiber 2004). It has also been shown that it is possible to synthesize diversity-oriented natural product-based libraries by chemical recombination of complex fragments obtained by chemical degradation of diverse bioactive natural products (Niggemann et al. 2002). REFERENCES
Copyright 2005 Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc05132f5.jpg] [oc05132f10.jpg] [oc05132f8.jpg] [oc05132f7.jpg] [oc05132f4.jpg] [oc05132f11.jpg] [oc05132f9.jpg] [oc05132f6.jpg] [oc05132f2.jpg] [oc05132f3.jpg] [oc05132f1.jpg] |
| |||||||||

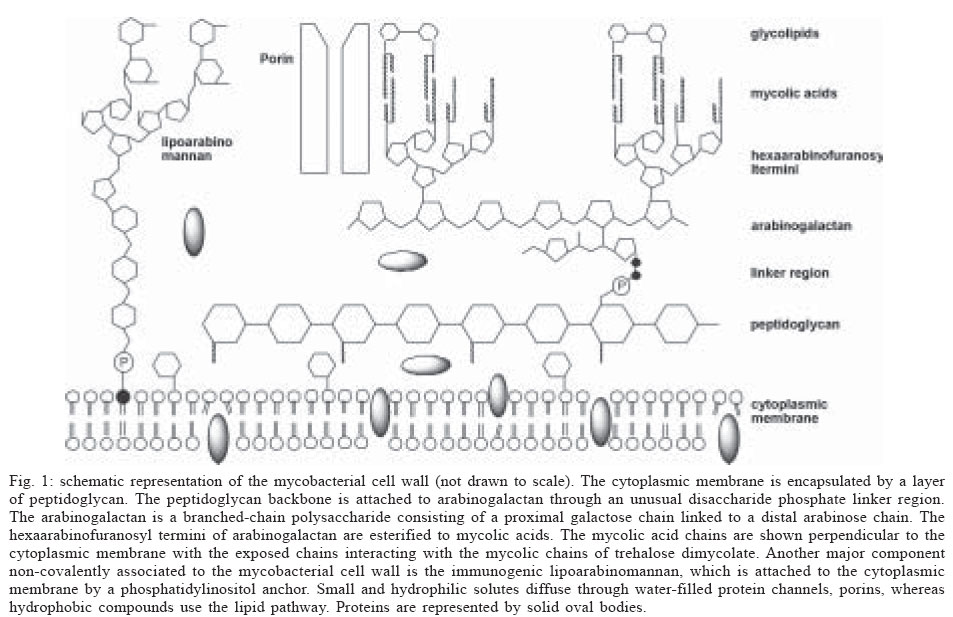{kind=link}
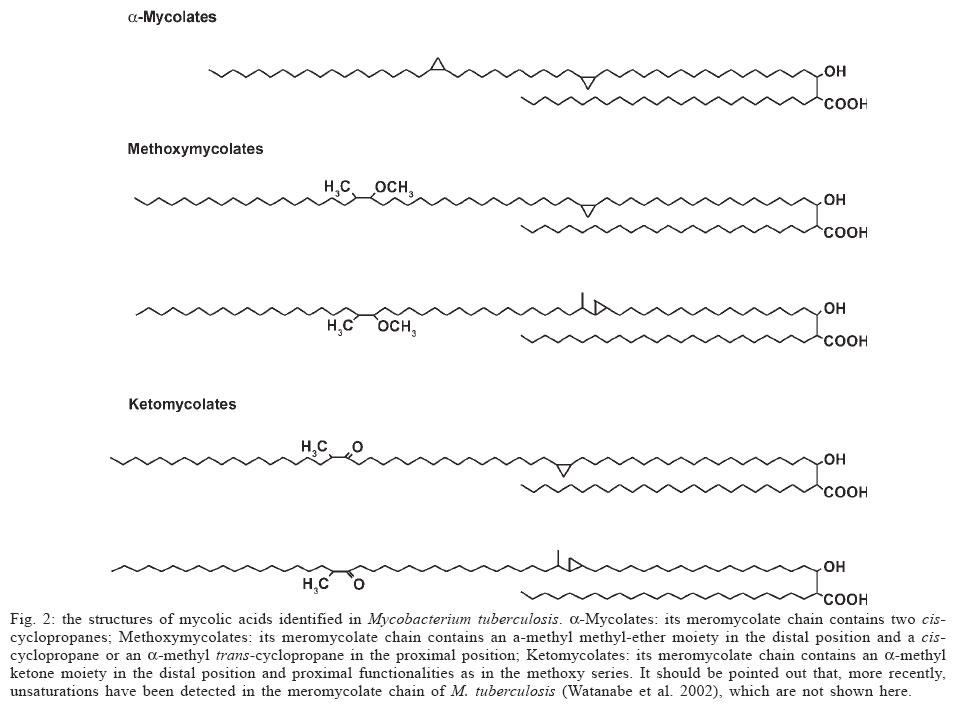{kind=link}
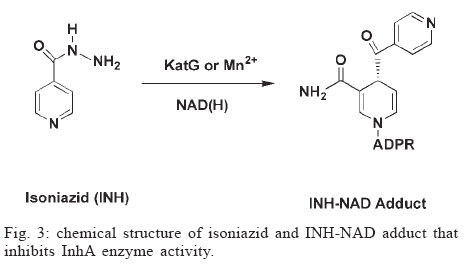{kind=link}
{kind=link}
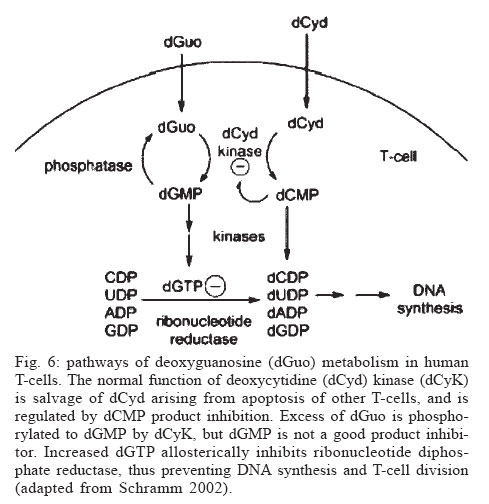{kind=link}
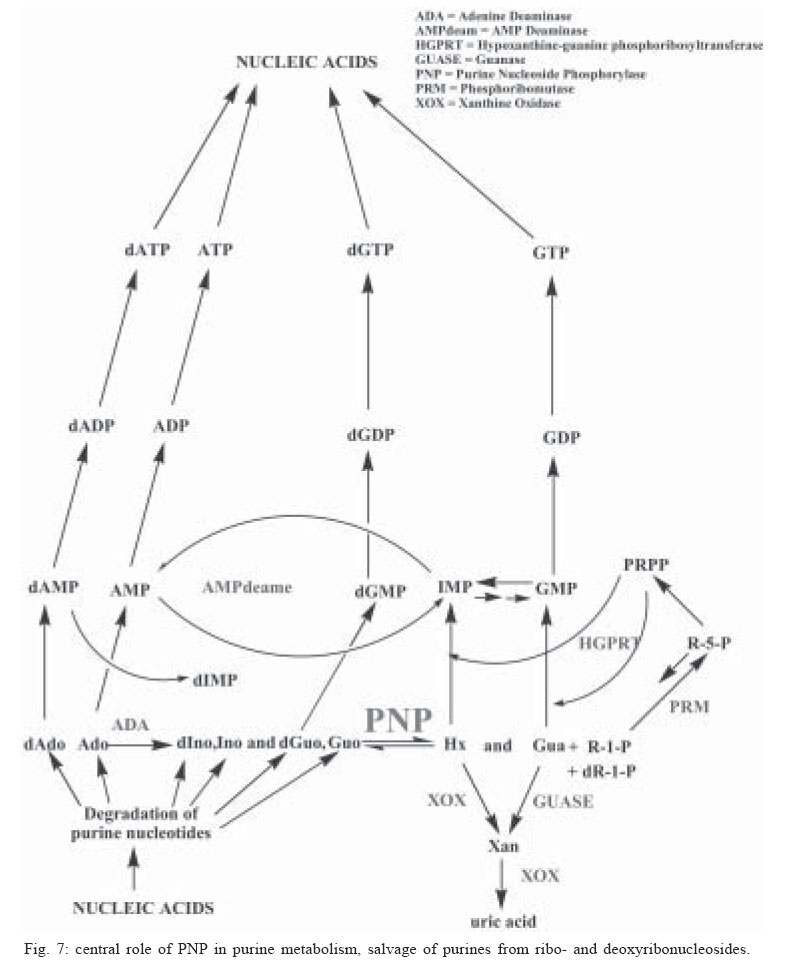{kind=link}
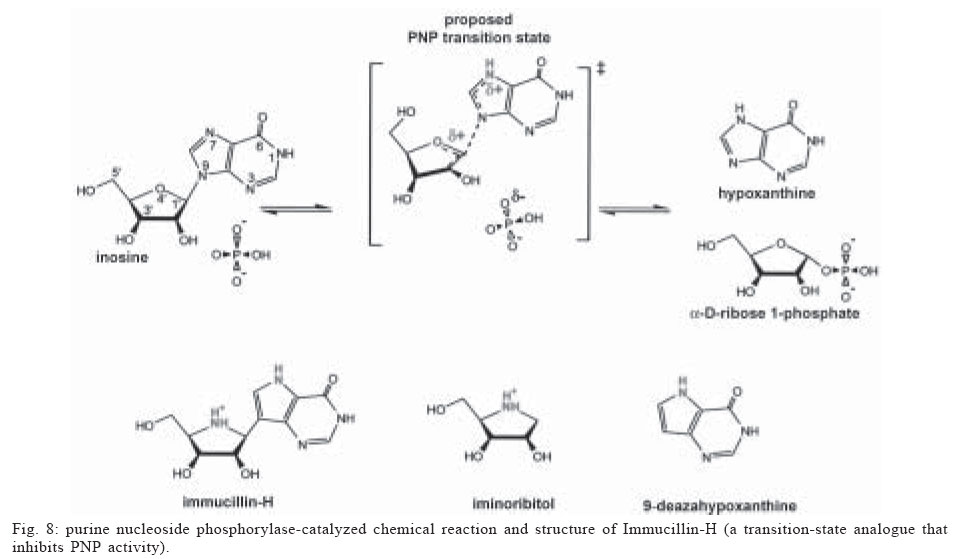{kind=link}
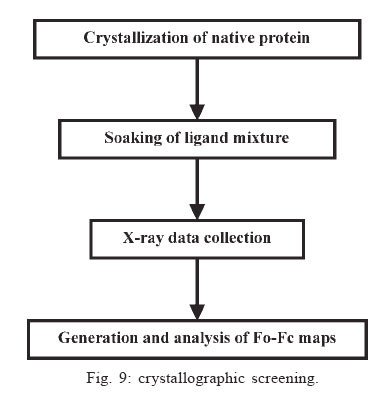{kind=link}
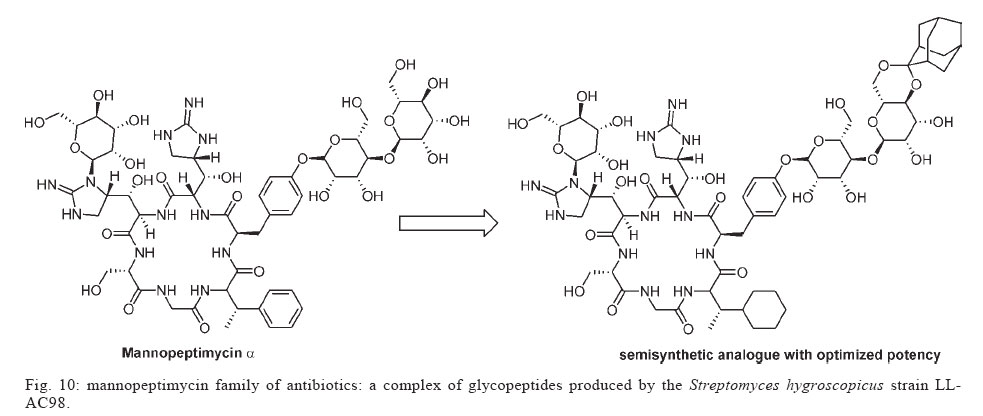{kind=link}
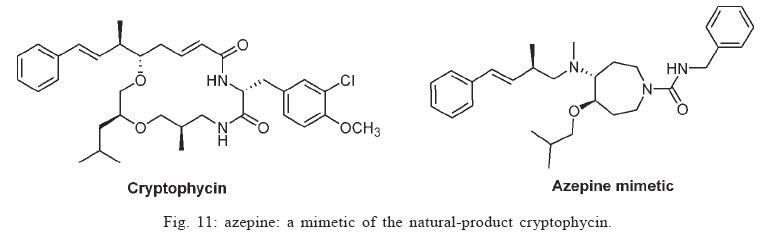{kind=link}