 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 101, No. 5, August, 2006, pp. 463-491 Evolution and pathology in Chagas disease - A Review Antonio RL Teixeira/+, Rubens J Nascimento, Nancy R Sturm* Laboratório
de Pesquisa Multidisciplinar em Doença de Chagas, Faculdade de Medicina,
Universidade de Brasília, Caixa Postal 04536, 70919-970 Brasília,
DF, Brasil *Department of Immunology, Microbiology and Molecular Biology,
David Geffen School of Medicine, University of California at Los Angeles,
LA, US Financial support: CNPq, Finep, FAP-DF 1 The manuscript Nitz et al. appeared in a July 2004 issue of Cell; this article was retracted by the editor in Sept 2005 (Retraction 2005, Marcus 2005) and thus can no longer be obtained from Cell as a legible transcript. Interested readers can obtain unadulterated copies of the original directly from ARLT. The authors stand by their work, analyses, and conclusions. At issue was the site of integration into the vertebrate genomes. We continue to challenge our conclusions experimentally (Simões-Barbosa et al. 2006), and aim to resolve any questions to the satisfaction of the scientific community. Received 16 March
2006 Code Number: oc06082 Trypanosoma cruzi acute infections often go unperceived, but one third of chronically infected individuals die of Chagas disease, showing diverse manifestations affecting the heart, intestines, and nervous systems. A common denominator of pathology in Chagas disease is the minimal rejection unit, whereby parasite-free target host cells are destroyed by immune system mononuclear effectors cells infiltrates. Another key feature stemming from T. cruzi infection is the integration of kDNA minicircles into the vertebrate host genome; horizontal transfer of the parasite DNA can undergo vertical transmission to the progeny of mammals and birds. kDNA integration-induced mutations can enter multiple loci in diverse chromosomes, generating new genes, pseudo genes and knock-outs, and resulting in genomic shuffling and remodeling over time. As a result of the juxtaposition of kDNA insertions with host open reading frames, novel chimeric products may be generated. Germ line transmission of kDNA-mutations determined the appearance of lesions in birds that are indistinguishable from those seen in Chagas disease patients. The production of tissue lesions showing typical minimal rejection units in birds' refractory to T. cruzi infection is consistent with the hypothesis that autoimmunity, likely triggered by integration-induced phenotypic alterations, plays a major role in the pathogenesis of Chagas disease. Key words: Trypanosoma cruzi - kinetoplast DNA - horizontal transfer - genome growth - kDNA-mutation - pathology Eons and interplay The enzootic known as Chagas disease or American trypanosomiasis (Chagas 1909, 1911) is presented here as a hallmark of the interplay among extant organisms from different taxon of increasing complexity united by circumstance. In the geological time scale of evolutionary history, play initiated with the undulipodia acquiring a nucleus (Lake & Rivera 1994), resulting from a radical revolution and a major discontinuity between the prokaryotic and eukaryotic organisms during the proterozoic, 1500 million years ago (mya) (Margulis & Sagan 2002). In the absence of fossil records, the history of protozoa has been written mainly by morphological and life cycle data. However, by the end of the twentieth century the availability of automated DNA sequencing enabled deduction of the evolutionary relationships of extinct species from the genomes of their extant relatives (Lake et al. 1988, Stevens et al. 2001). DNA sequence analysis necessitated the engineering of a molecular clock that permitted evolutionary dating (Douzery et al. 2003, Delsuc et al. 2004). The clock advances such that mutations underlying evolution behave as radioactive atoms, for similar reasons: the tautomeric shifts of purine and pyrimidine nucleotides, although an unpredictable stochastic process can be prognosticated with reasonable accuracy at regular intervals (Klein & Takahata 2002). In practice, however, some species accumulate mutations faster than others, and, like a grandfather clock, the molecular clock requires calibration based on the empirical sequence data from extant species. In addition, the Earth's development exerts an influence on biological evolution through subtle changes in climate and environment, or cataclysms such as tsunamis, necessitating adjustment of the clock to explain discontinuities beyond the Gondwanaland partition (Krause & Bonaparte 1993, Salgado-Laborieau 2001). However, chasms will remain wherever horizontal DNA transfer (HDT) among disparate species occurs (Nitz et al. 20041, Simonson et al. 2005, Babushok 2006), complicating reconstruction of the universal tree of life. The expected shifts (e.g., mutations) are essentially normalized, and DNA accumulates substitutions at a stable pace. Since mutations appear at more-or-less regular intervals, time can be estimated proportionally (Klein & Takahata 2002, Podlipaev et al. 2004). The constant accumulation of mutations affecting amino-acid change in proteins during evolution is thus accountable (Douzery et al. 2004). Molecular clock data used to reconstruct evolutionary history can be calibrated with congruent morphological and lifecycle data of existing organisms (Table I). The protoctist (Eukaryota, Excavata, Euglenozoa) ancestors of the protozoa are dated to the pre-phanerozoic (Margulis et al. 2000). Protozoa belonging to the Class Zoomastigophorea include the most interesting Order: Kinetoplastida. Included in the Family Trypanosomatidae are parasites of major medical and veterinary importance: Trypanosoma cruzi, which produces Chagas disease in the Americas, Trypanosoma brucei, the agent of sleeping sickness in Africa, and Leishmania species, responsible for leishmaniases worldwide. No clear division separates the Stercorarian trypanosomes and representatives have been found in lower vertebrates, such as the crocodile parasite Trypanosoma gray (Hoare 1972). Their closest relatives are the bodonids and cryptobiids that parasitize fish and amphibians (Donelson et al. 1999). Phylogenetic analyses based on small subunit ribosomal RNA (SSU rRNA) and heat-shock protein (Hsp90) first- and second-position codon nucleotides support placement of the root for the kinetoplastids next to the bodonids (Simpson et al. 2002), suggesting that trypanosomatids descended from the bodonids and that Boldo saltans is the closest extant relative. The interlocking network organization of kinetoplast DNA seen in trypanosomatids therefore is a derived condition from open-conformation minicircles found early in kinetoplastid evolution (Lukes et al. 2002). The molecular analysis of SSU rRNA to determine the phylogenetic relationship between Trypanosoma chelodina from tortoise (Emidura signata, Elseya latis-ternum and Chelodina longicollis) and Trypanosoma binneyi from platypus (Ornithorhyncus anatinus) excluded co-evolution of these trypanosomes with the vertebrate mammal host (Jakes et al. 2001). However, early evolutionary acquisition was achieved by exposing clean bullfrog tadpoles (Rana catesbiana) to leeches (Desserdobella picta) that had fed on frogs infected with Trypanosoma pipientis (Siddall & Desser 1992). The presence of trypanosomatids in the blood of aquatic invertebrates (leech) and vertebrates (fish) suggested that evolution of the former required secondary acquisition of host and habitat (Davies & Johnston 2000, Hamilton et al. 2005) during the phanerozoic, 570 mya. The evolution of the Stercorarian T. cruzi likely required stepwise adaptation to invertebrate and vertebrate hosts. A direct line of evidence showing a phylogenetic relationship between trypanosomatids of leeches, fish, and amphibians, and those of mammals cannot be drawn, but the close relationships between lizards and triatomines in an ecosystem located in Baja California, Mexico is suggestive. In this environment various triatomine bugs (Dipetalogaster maximus) and lizards (Sauromalis australis) dwell in burrows of exposed rocks in the absence of mammals. The complete T. cruzi lifecycle was observed in lizards that were infected by ingestion of the protozoan-infected D. maximus, and thereafter clean D. maximus acquired T. cruzi upon feeding from that lizard (reviewed in Teixeira 1987). The ultimate reservoirs of these trypanosomes may not be mammalian. The origin of multicellular animals as assessed through topology of 18S rRNA indicates that the Annelida-Mollusca are the closest relatives of arthropods. There are a million species of arthropod in the Class Insecta: Hemiptera. Using amino acids, nucleotides, and the mitochondrial molecular clock calibrated by Blattaria (cockroaches), Orthoptera (crickets and locusts), Hemiptera (true bugs), Diptera, and Lepidoptera (butterflies and moths), dates in accord with available insect fossils and biogeographical history were obtained (Gaunt & Miles 2002). The terrestrial transition of arthropod ancestors to insect ancestors coincided with the earliest vascular plant megafossil 434 to 421 mya, and the emergence of triatomine bugs occurred at 99.8 to 93.5 mya (Table I). By this date mammals harboring hemoparasitic trypanosomes presented the best intracellular niche for further differentiation and multiplication, thus fulfilling the current T. cruzi lifecycle growth requirements (Fig. 1). Hematophagy is the lifestyle of 14,000 insects dependent on the ionized iron [Fe++]-bound heme protein core in the hemoglobin molecule. The obligate hematophagy of triatomine bugs represents a primary factor in their biology, distribution, and evolution (Lent & Wygodzinsky 1979). The success of T. cruzi and of Triatomine spp. depends on the availability of [Fe++] in its environment, as limiting heme availability inhibits reproduction (Paiva-Silva et al. 2002, Maya-Monteiro et al. 2004), and, therefore successful adaptation resulted from this biochemical requirement of both partners. Among the hematophagous bugs belonging to the Family Reduviidae are the strictly hematophagous insects of the Subfamily Triatominae that became adapted to terrestrial ecoregions limited by parallels 42º in the Northern United States and 42º in Southern Argentina. The enormous diversification represented in the five families of triatomines (Carcavallo et al. 1997) has occurred within the major ecosystem habitats of the Americas (Dinerstein et al. 1995), fulfilling the specific bug's lifecycle requirements. South America's broad-leaf, humid tropical forests are a major dwelling of the branching tribe Rhodniini, mainly adapted to palm trees, whereas the tribe Triatomini adapted to rocky habitats and tree cavities (Lyman et al. 1999, Gaunt & Miles 2000), dwelling in the major dry ecosystems cerrado and savanna. Triatomines further adapted to specialist niches have the opportunity to select trypanosomes and mammalian hosts over the length of evolutionary history: In the palm niches dwell marsupials harboring trypanosomes defined as zymodeme 1 (Z1/DTU I), whereas in the tree cavities, ground burrows, and rocky outcrops are rodents and edentates (armadillos and anteaters) harboring trypanosomes of zymodemes Z2 and Z3 (DTU II subgroups a-to-e) (Yeo et al. 2005, Elias et al. 2005). Molecular clock phylogeny has suggested that genera Rhodnius and Triatoma branched off 40 mya (Gaunt & Miles 2000). At that time oral contamination was probably the most common route of infection of insectivorous mammals, including early primates. The interplay continued in the quaternary (less than 5 mya) when an enormous environmental ballroom enclosed mammal species belonging to seven orders distributed in 25 families (WHO 2002). These families comprise approximately 1150 extant species in the tropics (Patterson 1994), 80% of which are present in the Amazon Basin. Additionally, 20% of the small mammal fauna in certain Amazonian ecoregions are novel species beyond our reach (Silva & Patton 1998). There is no record of any mammalian refractory to T. cruzi infection. A 24 year-old polar bear (Ursus maritmus) died of acute Chagas disease acquired at the Guadalajara Zoo in Jalisco, Mexico (Jaime-Andrade et al. 1997). By contrast, unaccountably broad mammalian intra-specific diversity probably adds some glitter to the interplay and permissiveness to T. cruzi infection. These enzootic infections were probably common long before human speciation. Last minute The Tanzania Mumba Rock Shelter fossil of Homo sapiens sapiens has been dated to 0.13 mya (Brauer & Mehlman 1988). Radiation theory states that Homo erectus originated in Africa and migrated to other continents. Humans had developed primitive culture and considerable mental capacity before they reached the American continents. An alternative hypothesis holds that pre-historic Asians reached North America through the Bering Isthmus during glaciations 0.04 to 0.03 mya (reviewed in Salgado-Laborieau 2001). However, a human fossil located at Toca do Boqueirão, Serra da Capivara, in the northeast state of Piauí, Brazil, dates the arrival of Homo sapiens sapiens to the American continent 0.048 mya (Guidon & Delibras 1986, Bahn 1993). A third hypothesis supports the idea that Melanesians arrived in South America by boat (Salgado-Laborieau 2001). The Atacama Desert situated in Northern Chile and Southern Peru, possibly the driest place on Earth, was occupied by hunter-gatherer Amerindians 0.011 mya, at which time it was a route from the coast to the mountains (Neves et al. 1998). The environmental conditions in the desert (Berenguer et al. 1985) favored the conservation of mummified organic remains. Some specimens from desiccated human mummies showed the gross lesions typical of Chagas disease, and T. cruzi was identified by histological analysis (Rothhammer et al. 1985, Fornaciari et al. 1992). Tissue extracts yielded a PCR product that hybridized with a specific probe for the kinetoplast DNA of T. cruzi. The prevalence of Chagas disease in the Atacama Amerindian population reached 41% during a Holocene interval, ranging from 9,000 years to approximately the time of the Spanish conquest 500 years ago (Aufderheide et al. 2004). In this Andean region camelids and rodents were domesticated, and T. infestans had adapted to primitive human dwellings (Guhl et al. 2000). Thus, long before Colombus conquered the Americas T. cruzi infection was prevalent in the wild, and close proximity of triatomines increased the risk of secondary acquisition of the infection by humans. The five tribes of triatomines, including 130 species, are widely distributed from the Northern US to Patagonia in Southern Argentina (Carcavallo et al. 1999). At least 40 triatomine species can harbor T. cruzi, thus they are all potential transmitters of infection. Sympatry and syntopy, common features associated with transmission of flagellates to invertebrate and vertebrate hosts, are readily observed in the evolutionary history of T. cruzi infecting mammalian hosts in the Americas. The clock goes round Chagas disease was acquired easily by settlers of the Americas in the post-Colombus days. A disease that attacked newly-arrived individuals of European or African ancestry leading to sudden death or to a consumptive heart disease was known: A medical dictionary published in the 19th century registered "mal de engasgo", dysphagia related to Chagas mega-esophagus (reviewed in Teixeira 1987, Miles 2004). Nowadays, Chagas disease is hyperendemic in the regions of Latin America where the human population lives in close proximity to triatomines carrying T. cruzi. Countrywide surveys estimate that 25% of Latin Americans (100 million people) are at risk of contracting the disease, which today affects 18 million people (WHO 2002). On the basis of field studies conducted over the course of several decades, it has been estimated that 30% of the infected human population (5.4 million cases) will develop clinically overt disease. Mortality has been estimated at 0.56%, thus 100.8 thousand Chagas patients are expected to succumb to the disease per year (Prata 1975). The infectious agent and the host organism The trypomastigote forms of T. cruzi invade phagocyte cells at the site of entry in the body. Some of the invading flagellates may be destroyed, but many of the internalized parasites replicate and complete the cycle, differentiating into new forms that invade other cells and tissues. The intracellular amastigotes can persist dormant in the host body for decades, hidden in muscle cells without causing significant damage to the tissues. Target cell invasion involves microtubule-dependent lysosome recruitment and fusion at the site of attachment to the target cell plasma membrane (Tardieux et al. 1992, Andrade 2004, Chakrabarti et al. 2005). Upon internalization, the resulting acidic environment at the fusion site activates a secreted porin-like protein promoting trypomastigote escape from the phagocytic vacuole and differentiation into the dividing amastigote persisting in the cytoplasm (Andrews 2002) to initiate another cycle of infection. The fate of the infection depends on the ability of the virulent trypomastigotes to escape from the digestive enzymes in the phagolysosomes (Rao et al. 2004). Including a replicative lag period of at least 18 h and a mean population doubling time of 15 h, the complete intracellular cycle lasts four days (Engel et al. 1985). Cells harboring a high-density parasite load burst open and release differentiated forms that initiate new cycles of infection. Host defense mechanisms can control the reinfection process in moderate or low-density phagocyte invasion (reviewed in Teixeira et al. 1996). Although vertebrate monocytes and macrophages are remarkably effective in overriding microbial infections, some kinetoplastids have evolved to survive in phagocyte cells possessing active NADPH oxidases through contributions of respiratory burst-derived reactive oxygen (O2-, H2O2) or through nitrogen intermediates inducible by nitric oxide (NO) synthase (Murray & Nathan 1999). These mediators are related primarily to protective mechanisms. Hence, successful infection of macrophages depends on a variety of mechanisms that enable the brief existence of a flagellate within the harsh environment of a phagocytic cell (Andrews 2005). Conserved signal transduction pathways related to activation of glucose metabolism, energy consumption, protein phosphorylation, and oxidative burst activity play a major role in parasite-host relationships. The stimulus-response coupling through protein-kinase C (PKC), which is defective in mononuclear phagocytes, reflects attenuation of PMA-induced translocation of enzyme to the particulate fraction of infected cells (Olivier et al. 1992). The ability of flagellates to avoid protein kinases (MAPK)-, NF-kB, and extra-cellular signal-regulated kinase 1/2 activation of macrophages through a specific lipophospho-glycan-unit virulence factor is part of the strategy evolved to elude the host's innate defense. Significant modification in the phosphorylation status of tyrosine-containing protein in response to heat stress suggests that phosphorylation/dephosphorylation plays a role in signal transduction pathways for parasite entry and differentiation in the host cell (da Cunha et al. 2005). Various signal transduction events are triggered in the host cell during invasion by T. cruzi, which appear to regulate phosphatydilinositol 3-kinase (PI3K) and protein kinase B (PKB/Akt). Strong activity of PI3K and PKB/Akt was detected when macrophages are incubated with trypomastigotes or their isolated membranes, and this early invasion signal has been associated with successful parasite internalization (Todorov et al. 2000, Wilkowsky et al. 2001). The intracellular protozoan lifecycle appears to be regulated by phosphatidylinositol-specific phospholipase C and by protein tyrosine phosphates. Altogether, multiple defense mechanisms associated with protein phosphorylation networks play a major role in the control of T. cruzi growth and differentiation (Chakrabarti et al. 2005). A balance between these signal transduction events implies that parasite-cellular `cross-talk' exists regarding defense mechanisms and regulation of growth. Virulent T. cruzi isolates express an unusual family of glycoinositolphospholipids on their surface, closely related to glycosylphosphatydil anchors (Previato et al. 1990, 1995, Lederkremer et al. 1991, Almeida et al. 2000). These molecules fall into two categories, depending on the substitution of the third mannosyl residue in the conserved glycan sequence Man4-(AEP)-GleN-InsP04 by ethanolamine phosphate or b-galactofuranose (reviewed in Previato et al. 2005). The role of these molecules in promoting pathogenicity and parasite survival in the intracellular environment remains unknown. Interlocking networks The kinetoplast DNA (kDNA) contains a few dozen maxicircles (23 kb) and thousands of minicircles (1.4 kb) catenated into a complex network (Fig. 2A, B), comprising 10-15% of the total cell DNA (Lukes et al. 2002, Junqueira et al. 2005). Maxicircles encode the structural genes necessary for mitochondrial function (Westenberger et al. 2006), many of which are modified post-transcriptionally by a unique uridine insertion/deletion process called RNA editing (Simpson et al. 2004). In T. cruzi, each minicircle contains four equally-spaced conserved regions thought to contain origins of replication (Avliyakulov et al. 2003); the four intervening variable regions each have the potential to encode an individual guide RNA (Avila & Simpson 1995), the small transcripts that specify the location and number of uridines to be added to or deleted from the mRNA. The structure of the kDNA complex is not understood, nor have the intricacies of kDNA replication been fully elucidated. Replication involves doubling of the number of minicircles and maxicircles and distributing the progeny into two daughter networks that are each identical to the parent network (reviewed in Liu et al. 2005). Prior to their replication, minicircles are individually released from the network into the kinetoflagellar zone situated between the kDNA disk and the flagellar basal body (Ferguson et al. 1992). The proteins that initiate minicircle replication, including the universal minicircle sequence binding protein, primase, and replicative polymerase are localized within the kinetoplast flagellar zone (Li & Englund 1997, Abu-Elneel et al. 2001, Das et al. 2004). The revelation that minicircles are integrating into the host genome (Teixeira et al. 1991, 1994a,b, Simões-Barbosa et al. 1999) and correlated to the pathology of Chagas disease (Nitz et al. 2004) has renewed interest in understanding the composition of minicircles within the known sub-groups of T. cruzi. Minicircle kDNA integrations behave as mutagens in the host. kDNA has been used for the identification of subgroups of T. cruzi populations (Avila et al. 1990). The relationships among distinct parasite populations have been characterized using largely nuclear markers into six discrete typing units (DTUs) (Brisse et al. 2000a,b, 2001). The definition of these DTUs allows a systematic approach to the analysis of kDNA in order to assess any direct links with pathogenicity, and three clades have been defined based on maxicircles sequences (Machado & Ayala 2001), however the minicircle population dynamics that could vary from isolate to isolate. Genetic exchange and diversity So it is necessary, at least intermittently…, this thing called sex. As of course you and I knew it must be. Otherwise surely, by now, we mammals and dragonflies would have come up with something more dignified (Quammen 1985) The sexual lifecycle may reflect the history of life's programming and adaptation to an oxygenated biosphere. Sex is an ancient cellular capacity present in primitive eukaryotes. So vital, it may have originated with an advantageous symbiotic union (Margulis & Sagan 1995) of two bacteria with complementary metabolic strategies. Many organisms thought to be exclusively asexual also reproduce sexually (Maynard-Smith 1998). Continuous refinement of this magnificent engineering results in an unresolved problem, a missing link to sexual fertilization (Redfield 1999, Jan et al. 2000). The contribution of a-karyotic genomes and membranes in the evolution from two pro-karyotes to one eu-karyote (Margulis et al. 2000) is evident, with each gamete sharing common descent with extant microbial species in two moneric domains: oocyte to eu-bacteria (since eukaryotes have eubacterial membranes), and sperm to archea (Walther et al. 1999). It seems likely that the sexual reproduction process used by early prokaryotes was a prerequisite to large evolutionary leaps through cycles of cell fusion and chromosome segregation, possibly providing selective recombination advantages to molecular parasites. Character compatibility of molecular markers used to distinguished asexual and sexual reproduction represents a potent tool (Mes 1998) complementary to the use of high variability dominant markers such as random amplified polymorphic DNA fragments (RAPDs) and amplified fragment length polymorphisms in genetic analyses of trypanosomatids. Genetic exchange is now known to occur during the life cycle of many parasitic protozoa, including trypanosomes. Among the African trypanosomes, crosses between T. brucei individuals resulted in hybrids formed during tsetse fly transmission. The hybrids appear mainly diploid, but various traits in some chromosomes were inherited in a non-Mendelian fashion (Walliker 1989). Isoenzyme and RAPD analyses of T. cruzi isolates from Central and South America showed two homozygous and the corresponding heterozygous phenotypes consistent with genetic exchange (Carrasco et al. 1996). Similarly, two T. cruzi stocks that carried different drug-resistance markers were co-passaged through an entire life cycle. Six double-drug (hygromicin and neomycin) resistant trypanosomes were recovered from the mammalian stage of the lifecycle, showing fusion of parental genotypes, loss of alleles, homologous recombination, and uniparental inheritance of kDNA. These results are consistent with hybrid genotypes among natural isolates of T. cruzi that show aneuploidy and recombination across vast genetic distances, consistent with non-Mendelian genome duplication (Gaunt et al. 2003). An analysis of multiple nuclear markers has led to the proposal (Fig. 3) that the extant lines of T. cruzi can be described by a grand total of two such genetic exchange events (Sturm et al. 2003, Westenberger et al. 2005), which are consistent with the distribution of the three clades defined for the maxicircle lineages (Machado & Ayala 2001). The demonstration of genetic recombination resulting in marked polymorphisms requires the use of appropriate nomenclature. Here the term stock rather than strain will be used to designate the wildtype T. cruzi isolates. This term is in keeping with the fact that T. cruzi stocks kept in the laboratory will not maintain stable physiological and biochemical markers after serial passage in vitro and in vivo. Since T. cruzi populations show broad plasticity due to the capacity to transpose lineage, the word `clone' will not be used to designate the progeny of a single T. cruzi individual either, because over time the identity of the ancestor fades. Polymorphisms of T. cruzi populations isolated from Chagas heart patient hSLU239 and cloned subpopulations h1 and h2 and from Chagas megacolon patient mSLU142 and cloned subpopulations m1, m2, m3 and m4 were studied. Subpopulations h1 and h2 differed from those derived from the subpopulations m1-m4 in some 13 enzymes analyzed. RFLP analysis showed polymorphisms in four glycolytic enzymes and separate the T. cruzi stocks and their subpopulation isolates into three groups: I, formed by hSLU239 and isolate m4, which was classified as homozygous CC and BB. II, composed by mSLU142 and isolates h1 and h2, which was classified heterozygous for aldolase; III, including isolates m1, m2, and m3 classified as homozygous for each of five enzymes. Thus T. cruzi infection in each Chagas patient is produced by genetically diverse, polymorphic parasite populations (Lauria-Pires et al. 1996). Furthermore, the particular growth kinetics, doubling time and differentiation in axenic liquid medium of each of those parental stocks and derived subpopulation isolates revealed broad behavioral diversity (Lauria-Pires et al. 1997). These observations are consistent with several studies (Dvorak 1984, Engel et al. 1985) showing significant inter- and intra-group differences between growth rates, varying amount of total DNA/parasite subpopulation and marked heterogeneity of parasite populations to the intracellular cycle of T. cruzi. Virulence and pathogenicity are associated with diversity of parasite stocks and isolates. Neither the kinetics of growth, doubling time, differentiation in axenic culture, zymodemes, nor the restriction fragment length polymorphism assays showed correlations with parasitemia and pathogenicity in mice and, therefore, clinical and pathologic manifestations of disease could not be associated with intrinsic characteristics of T. cruzi populations (Lauria-Pires & Teixeira 1996). Moreover, to determine the role of T. cruzi superinfection on the outcome of disease, groups of BALB/c mice were prime-infected with low virulence isolates h1 and h2 and challenged with high virulence isolates m3 and m4. All mice injected with the m3 and m4 isolates succumbed before or at 16 days post-infection. In contrast, all mice injected with the h1 and h2 isolates survived the prime infection and were super-infected with the m3 and m4 parasites. Low-level parasitemias were observed after challenge with virulent parasites, and the histopathological lesions and mortality ratios were not different from those seen in mice that received a single T. cruzi injection. Thus morbidity and mortality in BALB/c mice infected with T. cruzi subpopulations are not associated with the frequency or parasite burden (Lauria-Pires & Teixeira 1997). The protective effect of primary infection with non-virulent T. cruzi subpopulations against subsequent challenge with virulent parasites was determined in groups of BALB/c mice. Low degrees of parasitemia were observed in mice challenged with the highly virulent clones and their survival ratios were not different from those seen in mice that received a single injection of non-virulent T. cruzi (Lauria-Pires & Teixeira 1997). Regardless of the intra-group genetic diversity, infection with non-virulent T. cruzi prevented severe infection in mice subsequently challenged with highly virulent parasites. Acquired immunity to T. cruzi infection The basis of acquired immunity to T. cruzi has been reviewed extensively (Teixeira 1987, Brener & Gazinelli 1997). Thus, we revisit the protective immunity concomitant to T. cruzi infection in mammal hosts, which bears features resembling those described for other intracellular chronic infectious diseases such as leishmaniasis, tuberculosis, and leprosy. Initial infection may initiate unperceived by the hosts in a large majority of cases and full-blown acute infections are occasionally seen in children below 15 years of age. Regardless of the mode of initiating invasion of the host organism, the infection persists in the body life-long, albeit with a continuously decreasing parasite load. As infections progress to the chronic stage they tend to become cryptic and demonstration of the infectious agent becomes progressively more difficult, although its presence can usually be confirmed by immunologic and molecular assays. The ongoing biological process follows a course that favors species survival in a great majority of cases: The host-parasite relationship persists with the absence of disease manifestation, and the host usually dies of causes unrelated to Chagas disease. The roles played by host humoral and cell-mediated immune reactions are crucial in disease outcome. We discuss aspects of host resistance not reviewed previously. CD4+ Th 1 lymphocytes are the main conducters for induction of partially protective immunity against T. cruzi (Hoft et al. 2000). CD8+ T lymphocytes, interferon-(INF)γ, and macrophages are important elements controlling parasite replication during the acute phase of infection. In the chronic stage of the disease, parasite-specific antibodies that fix complement and lyse the blood trypomastigotes are thought to be the main effector molecules to secure latency of infection. LTh type 1 cells that secrete inter-leukin(IL)-2 and γ-IL seem to be involved in Tc-, macrophage-, and IgG2-producing cells. LTh type 2 secreted IL-4, IL-5, and IL-10 are implicated in the humoral immune response and inhibition of LTh 1, Tc and macrophages (O'Garra & Murphy 1993). The depletion of CD8+ cells before inoculation resulted in increasing parasitemia and mortality in mice. Knock-out of CD4 and CD8 genes augmented the inflammatory response and parasite release in affected tissues. In inflammatory infiltrates CD4_ and CD8_ lymphocytes were seen (Sun & Tarleton, 1993). T. cruzi inoculation in resistant and susceptible mice showed that LTh cells are critical in determining important features of the protective immune response. The dominant TLh1 response was associated with resistance mediated by LTc and γ-INT that induce the macrophage L-arginine metabolic pathway and free radical production. LTc lymphocytes were associated with elimination of T. cruzi-infected host cells and with inflammatory heart lesions (Laucella et al. 1996). Nevertheless, Martin & Tarleton (2005) have observed vigorous anti-parasite response from both CD4+ and CD8+ T cells. It was shown that mice with variant natural killer T (NKT) cells bear striking inflammatory infiltrates of dendritic, natural killer, B, and T cells. These inflammatory cells produce increasing concentrations of gamma interferon, tumor necrosis factor alpha and NO, show a diminished antibody response, and usually die. A subpopulation of invariant NKT cells appeared to dampen the inflammatory response and mice survival. The pivotal role of IL-12, γ-INT, and iNOS in the control of parasitemia, inflammation, and host resistance is antagonized by the IL-4 (Michailowsky et al. 2001). NKT cells play a role in the regulation of the immune responses during infection and autoimmune disease (Duthie & Kahn 2005). In human Chagas heart disease there is a predominance of CD8+ lymphocytes, which are many-fold more frequent than CD4+ T cells (Reis et al. 1993). In the gastrointestinal form, however, a significant decrease in the absolute number of CD3+ lymphocytes and in CD19+ B lymphocytes as well as an inversion of the CD4/CD8 ratio was seen, whereas this ratio remained unchanged in the heart patients as compared to that of control (Lemos et al. 1998). These T cells showed down-regulation of CD62L, which has been associated with effector memory phenotype. Further characterization of gene expression in Chagas heart disease showed immune response, lipid metabolism, and mitochondrial oxidative phosphorylation genes selectively up-regulated. The γ-INT signaling pathway up-regulating chemokines could be involved in heart hypertrophy (Cunha Neto et al. 2005). Regulation of the partially protective immune response in naturally infected humans and in experimental T. cruzi infection in mice is not understood precisely (Marino et al. 2005). Partially protective immune responses associated with host resistance to T. cruzi infection may be involved with severe inflammatory infiltrates and pathology in the heart, digestive tract, and other tissues (Correa-Oliveira et al. 1999, Gomes et al. 2003). Thus, these studies contraindicate the prospect for a successful vaccine, despite enthusiasm for the effort (Levin 1996, Kierszenbaum 1999). Clinical presentations of Chagas disease Key practical points in human and veterinary medicine are those associated with emerging symptoms and signals that can be recognized as clinical features of a disease. Any persistently infectious biological process can be divided into as many segments as required for facilitating measures that leads to palliation of symptoms and treatment of signals. T. cruzi infections in mammalian hosts have been divided into successive acute and chronic stages. Acute Chagas disease Most cases of acute T. cruzi infection are ascribed to triatomine bugs, the insect vector that transmits the protozoan. A delayed-type indurate skin lesion may appear at the portal of parasite entry in immune-competent hosts, but not in immune-compromised patients lacking cell-mediated immune response (Teixeira et al. 1978, Teixeira & Teixeira 1995). Acute infection in the latter case goes unperceived by the patient and/or by the physician in the absence of diagnostic signs and symptoms. A field study showed that approximately 75% of the acute cases were seen in children less than 10 years of age (reviewed in Teixeira 1987). Death in acute Chagas disease patients (possibly one case in 1000 acutely infected) is usually related to heart failure and/or meningitis and encephalitis. Sinus tachycardia, first-degree AV block, low QRS voltage and primary T wave changes can be recorded by electrocardiograph (ECG). X-rays in such cases show an increased cardiac silhouette of varying degrees. Interestingly, all symptoms and signals that correlate with irrevocable involvement of different organs cede spontaneously without apparent sequel (WHO 2002). Indeterminate phase The chronically infected individual remains a life-long source of the parasite as an indeterminate phase reservoir. Approximately one third of all individuals with indeterminate infections will develop chronic Chagas disease. The indeterminate phase of infection has been defined based on criteria of (i) positive specific IgG antibody test and/or parasitological demonstration; (ii) absence of symptoms and signs of Chagas disease; (iii) absence of ECG abnormalities; (iv) regular size of heart, esophagus and colon by X-ray. Using these criteria, regardless of the geographic area where field studies have been conducted, it has been estimated that two thirds of T. cruzi-infected individuals will remain in this condition during their economically-productive lives. Thus most patients with indeterminate infections are 20 to 50 years of age, comprising approximately 12 million people with positive immunologic tests for the parasite. Their life span is similar to that observed for the general population (Macedo 1999). Usually indeterminate phase individuals are identified as such during job application or blood bank screening. Denial of work is unjustified, but the candidate is disqualified from blood donation. Chronic cardiac disease The cardiomyopathy associated with chronic infection has made Chagas disease the most lethal endemic infectious disease in the Western hemisphere (Cubillos-Garzon 2004). In a randomized urban study, the prevalence of chronic Chagas disease reached 18% of the street cleaners Brasília, Brazil (Lauria-Pires et al. 2000). Among these 245 chagasics, only two could recall an acute infection stage. This description is in keeping with the epidemiological truth that for each chagasic with a clinically disclosed acute phase of the infection, there are 125 cryptic patients. Clinical studies have shown unparalleled levels of ECG abnormalities in patients showing positive serologic tests for infection when compared with the non-chagasic population. The main life-threatening manifestations of chronic heart disease are heart failure, arrhythmias and thromboembolism. ECG abnormalities are cumulative over time and become more frequent from 20 years post acute infection. Chronic T. cruzi infections lead to an increasing number of ECG alterations recorded on two occasions 10 years apart (Lauria-Pires et al. 2000). Ventricular premature contractions, right bundle branch block, combined branch block, intraventricular conduction disturbance, and ventricular repolarization changes were recorded more frequently over time. These alterations were many-fold more frequent in Chagas patients than in controls (p < 0.001). The progressively shifting right bundle branch blocks in particular were recorded more frequently than the remaining ECG alterations in the Chagas patient population. ECG revealed ventricle wall hypokinesis and intracavitary thrombus. The heart enlargement in Chagas patients is an ominous sign, leading to a poor prognosis (Lauria Pires et al. 2000). A word of caution is needed because the severely compromised Chagas patient may die during a Holter-type ECG monitoring of arrhythmias and other heart disturbances. Moreover, patients having apparently stable ECG changes may show sudden flare-ups of an underlying defect leading to heart failure. The variability of the ECG and clinical manifestations is remarkable: Some patients showing cumulative changes in successive ECG recordings apparently halt the evolving life-threatening arrhythmias and lead a normal life. In one population of chronic patients, 57% mortality was related to Chagas disease, of which 58% had heart insufficiency, and 37.5% died suddenly (Prata 1999). The remaining deaths were related to the digestive forms associated with chronic disease. The average time between acute infection and development of severe chronic Chagas lesions has been estimated at 28 ± 7 years (Prata 2001). However, Chagas cardiomyopathy rapidly progresses toward death no longer than 5 years after signs of heart failure. A common finding in death resulting from heart failure is cerebral infarction as a consequence of a thrombus detached from the left ventricle. Digestive form of Chagas disease Gastrointestinal disorders are among the most common clinical manifestations of chronic Chagas disease. A clinical study aimed at the evaluation of autonomic function in chagasic patients showed esophageal alterations usually occurred earlier in the course of the chronic infections when compared to similar abnormalities in the heart (reviewed in Macedo 1999). A digestive disease patient may complain of difficulty swallowing and regurgitation of food, clinical symptoms related to megaesophagous. Alternatively, some complain of constipation due to a fecal bolus in the rectal ampoule as a result of megacolon. Megaesophagous and megacolon are seen frequently in endemic areas; patients showing these conditions bear specific antibodies to T. cruzi and consistently positive nucleic acid tests (NAT) assays. These `mega' conditions can affect individual patients independently, in association with each other or in association with heart disease. Megaesophagus can manifest clinically in chagasics as early as 2 years of age or any time throughout life, although the majority of cases are seen in men between 20 and 40 years old. Megaesophagus anticipates heart trouble in many patients. The disease manifests by dysphagia, heartburn, hiccups, regurgitation of food, and increasing salivation. This clinical picture associates chagasics megaesophagus and caquexis resulting from difficulty to eat. The disease appears to evolve during periods of dysphagia followed by long periods during which symptoms are absent. Megacolon is seen considerably later in the course of Chagas disease in comparison with megaesophagus. The main symptom of chagasic megacolon is constipation. The progressive retention of hardened bolus leads to dilation and thickening of the walls of the colon, usually compromising the sigmoid colon and rectum. Difficulty with passing of bolus leads to dilation of the remaining intestines, increasing bowel movements, pain, and constant physical distress. The long-term use of laxatives can cause ulcerations of mucosal surfaces, septicemia, rupture of the wall of the intestine and peritonitis. Typical complications of megacolon are intestinal obstruction and rupture (reviewed in Prata 1999). Pathology in human Chagas disease Data on gross and microscopic pathology of acute Chagas disease was derived from two cases (Teixeira et al. 1970). The access to slides with sections from the two patients was kindly provided by Dr Moysés Sadigursky, from the Hospital of the Federal University of Bahia, Brazil. Data on chronic Chagas disease stems from ARLT's records of 20 post-mortem studies carried out personally at the Hospital of the Federal University of Bahia, Brazil, and at the Hospital of the University of Brasília, Brazil. Acute Chagas disease In the human body T. cruzi can parasitize any tissue derived from embryonic mesoderm, endoderm, and neuroectoderm. However, the intensity of infection in the body varies from case to case, likely depending on the genetics of host and parasite (Campbell et al. 2004). Mesoderm-derived conjunctive tissues smooth and striated muscles, bone marrow and phagocytic mononuclear systems, and gonadal cells can be heavily parasitized. The histopathology of an 18 month-old boy and of a 4 month-old girl who died of acute Chagas disease (Teixeira et al. 1970) revealed niches of amastigotes inside the theca cells of the ovary and inside the goniablasts of seminiferous tubes of the testes. Reproductive apparati were rarely examined previously in the course of T. cruzi infections. Endodermal tissue structures are eventually parasitized by amastigotes; epithelial cells of liver, kidneys, thyroid, pancreas, and other glands are spared infection. Neuroectoderm cells are parasitized less frequently than other embryonic-derived tissues; if the infection reaches the nervous system, glial cells, usually astrocytes, are invaded. Some T. cruzi isolates concentrate infection in the mononuclear phagocytic system, while others distribute themselves randomly in non-phagocytic muscle cells, where they evade the immune system. In striated heart and skeletal muscle amastigotes form large niches (a pseudo-cyst or cavity without a limiting wall) in the absence of inflammation. However, degenerative features of parasite-free muscle cells can be associated with inflammatory infiltrates. Similar aspects are present in smooth muscle cells all through the digestive tract. Features of the inflammatory lesions in the digestive tract are similar to those in the heart in that mononuclear cells invade parasympathetic ganglion structures situated between the peripheral and inner layers of the walls (Auerbach's plexus) and in the internal sub-mucosal layer (Meissner's plexus). Amastigotes may be present in fibroblasts, Schwan cells, and glial cells, but not in neurons. Nevertheless, neuronal lysis associates adherence of mononuclear cells of the immune system and depopulation of target neurons. Inflammatory infiltrates associate with glial and neuronal cells, secondarily compromising parasite-free neurons. Typically the heart of a deceased acute chagasic patient is enlarged, flabby, dilated, and congested. Lymph nodes situated between the aorta and the pulmonary artery, are engorged. The epicardial surface shows wide-open coronary vessels accompanied by whitish lymphatic vessels with tiny pearl-like nodules. These morphological changes presage the striking inflammatory infiltrates running through the walls of the heart. Microscopically, many muscle fibers and occasionally interstitial histiocytes show nests of dividing amastigotes. Mononuclear cells, mainly small and large lymphocytes with expanded cytoplasmic processes, and macrophages infiltrate the myocardium and adhere to the membrane of target heart cells. Several aspects of characteristic target cell destruction can be observed. Notably, some parasitized cells are isolated from the destructive inflammatory infiltrates. Parasite-free heart cells are mostly rejected or destroyed by immune system mononuclear cells. Confluence of multiple rejection units comprises the overall microscopic picture of lesions in acute Chagas disease. Inflammatory infiltrates invade cardiac parasympathetic ganglia where glial or Schwann cells can be parasitized, but neurons are spared. Adherence of inflammatory mononuclear cells to neurons leads to lysis and loss of several of these neural units in the acute phase. Additionally, inflammation extends into sympathetic nerves in the epicardial and intramural heart structures. Involvement of central nervous system structures in the acute phase can be frequent, as T. cruzi can be recovered from the cerebral spinal fluid in 72.7% of cases (Hoff et al. 1985). However, in half of these cases there is a lack of alteration of fluid components and absence of neurological damage. In acute cases showing clinical manifestation of neurological involvement, lesions are related to meningitis and encephalitis. Overall, the brain can show congestion of blood vessels and edema. Brain tissue is damaged sparsely by inflammation around small blood vessels, vascular hemorrhages, and microglial cell nodular proliferation in the white and gray matter. Typically inflammatory cells invade the meningeal leaflets and encircle blood vessels inserted deep in the brain. T. cruzi nests can be seen in brain astrocytes. Indeterminate phase Microscopic substrates for changes in the indeterminate phase were drawn from 20 biopsies revealing minimal inflammatory heart lesions (Mady et al. 1982) that were generally focal and small. Skeletal muscle biopsies showed spotty inflammation, target cell lyses and degeneration (Sicca et al. 1995). Inflammatory lesions in the heart, digestive tract, and skeletal muscle are similar to those seen in clinically manifest chronic disease patients, but to a much lower degree. Inflammatory infiltrates surround muscle fibers, resembling the minimal rejection unit of the target heart cell. In some cases multiple rejection units compromise a bundle of fibers in a single muscle; any nerve or sympathetic ganglion in the region can be affected by inflammatory infiltrates. In the digestive tract, lesions reaching the parasympathetic ganglia and neuronal cell depopulation have been observed (Lopes 1999). Progressive clinical-pathological lesions present in chronically infected chagasics classify the disease according to the affected organ. Chronic Chagas heart disease Chronic Chagas heart disease affects individuals of both sexes equally, usually between 30 and 45 years of age. In patients showing progressive ECG alterations unexpected deaths occur at a rate of 37.5% (Prata et al. 1986, Lopes 1999), whereas 58% develop ominous signs of heart failure and usually die within 7 to 24 months (Dias 2000). Congestive failure involves the right and left chambers of the heart, thus affecting all blood circulation. The heart increases in size, occupying the base of the thoracic cavity and bulging against the chest wall. The average heart weight reaches 540 ± 90 g, in patients dying of congestive failure, whereas in those undergoing sudden death the weight reaches 390 ± 50 g. At the endocardial surface, the chambers of the heart become dilated and the walls can be thickened. A typical gross feature is the effacement of the apex of the left ventricle, showing aneurismal dilation. The presence of thrombus at different stages is seen frequently in the apex of the left ventricle and in the right atrium, and can be associated with thrombus embolism in the lung, brain, spleen, and kidney. Thrombus embolic phenomena in the brain and lungs are associated often with the ultimate cause of death in chronic heart disease. The epicardial surface of the heart shows dilated coronary vessels accompanied by lymph vessels with periodic, small, pearl-like nodules indicative of the drainage system from the subjacent myocardium inflammatory process. The main microscopic findings in the heart of a patient dying of Chagas disease are related to inflammatory infiltrates that are present in every case. The presence of parasitized heart cells can be detected microscopically in a small number of cases (10-20%), thus confirming NAT assays showing remnants of parasite nuclear DNA (nDNA) in affected tissues (Braga et al. 2000, Lauria Pires et al. 2000). The presence of parasite nests is seen more commonly in areas of the myocardium spared from inflammatory infiltrates. The infiltrates of macrophages, small and large lymphocytes branch from lymphatic vessels off the interstitial conjunctive to surround muscle fibers. At inflammatory sites other cells can be present to a variable degree, including plasma cells, neutrophils, eosinophils, and mast cells. Ganglionitis, neuron degeneration, and neuron drop-outs are unique pathological findings in Chagas heart disease. At lesion sites, lymphocytes encircle target parasite-free target cell outer membranes, invade their cytoplasms, and induce lysis of the cell. A typical lesion showing a minimal rejection unit of a heart myofiber is circled in. The confluence of numerous rejection units induces severe diffuse myocarditis and accompanying changes in heart structure. Destroyed myofibers are replaced by fibrous tissue in the presence of evanescent inflammatory infiltrates. Inflammatory cells infiltrate specialized myofibers of the heart conductive system in the same manner that they infiltrate contractile myocardium. The intensity of this self-destructive inflammatory process varies from site to site in the myocardium; when some lesions are initiated, others are intermediate or phasing out. Some areas of the heart may be spared while others are severely damaged by inflammation; the intensity of these processes reaching the entire heart simultaneously would be fatal. At the ultra-structural level, in addition to association of mononuclear infiltrates with target cells undergoing lysis, myofibers show features of hypertrophy, mitochondrial swelling, necrosis, hyaline degeneration, disruption, and loss of myofibrils (Tafuri et al. 1973). As lesions increase in age loose fibrous tissue and inflammatory cells are replaced by dense fibrous scars that can be seen scattered throughout the heart walls (Rossi 2001). Chronic Chagas mega syndromes Pathology of the esophagus and colon associated with chronic Chagas syndromes is dependent essentially on inflammatory lesions upon smooth muscle fibers of the walls of the hollow viscera in the digestive system, affecting particularly the intramural parasympathetic neurons. Cases of mega syndromes affecting the stomach, duodenum, gall bladder, vesicle bladder, and bronchus have been reported also (Adad et al. 2001). For each of these conditions the pathology shows a common denominator described for most prominent esophagus and colon conditions (Hagger et al. 2000): Inflammatory lesions in parasympathetic ganglia lying between the smooth muscle layers (Auerbach's plexus) and in the sub-mucosal (Meissner's plexus) of the hollow viscera lead to ganglionitis, and neuron drop-outs as described for intracardiac ganglia. Although the presence of parasite nests in peri-ganglion fibroblasts or intra-ganglion glial cells has been reported, there are no accounts of parasympathetic neuron invasion (Tafuri 1971, Da Mata et al. 2000), nor is there evidence for a hypothesized neurotoxin secreted by the parasite (Andrade & Andrade 1966) and, therefore, neuronal depopulation does not correlate with parasite-induced host cell death. To the contrary, neuron death is associated clearly with immune system mononuclear cell adherence and lyses, as for that lesion type the minimal rejection unit correlates with destruction of heart target cells in Chagas disease. Active ganglionitis resolves with neuronal depopulation and fibrous tissue replacement. In summary, the most conspicuous lesions in Chagas mega syndromes parallel those in the heart and associate inflammatory mononuclear cell infiltrates with the minimal rejection unit that appears to be a common pathological denominator in human Chagas disease. The comparative pathology of Chagas disease At some stage, blood-sucking invertebrates (leeches) feeding upon fish, amphibians, and reptiles, acquired vertebrate trypanosomes and subsequently passed them on to terrestrial birds and mammals. Trypanosome infections in aquatic vertebrates could improve our understanding of the host-parasite relationships (Cox & Moore 2000, Hamilton et al. 2005). The absence of a fully-developed immune system in aquatic vertebrates could correlate to some extent with their lack of symptoms or minor pathogenic manifestations. This area demands further exploration, potentially leading to establishment of the ontogeny of the innate mechanism of resistance and/or susceptibility of vertebrate hosts to trypanosomatids parasites. Birds Birds are refractory to T. cruzi infection. Upon intravenous or intramuscular injections T. cruzi disappears immediately from the site of inoculation (Teixeira 1987) and cannot be recovered from bird blood by any means. However, inoculation of T. cruzi infective trypomastigotes in the air chamber of fertile chicken eggs results in intracellular growth of parasite amastigotes in embryo cells until the 10th day post-fertilization. Thereafter, the infection is eliminated by the embryo's innate immune mechanism (Nitz et al. 2004). Host-specific trypanosomes of birds can produce severe infections (Chandenier et al. 1988, Sehgal et al. 2001). Avian-specific Trypanosoma bouffardi experimental infections produce severe pathology in canaries. Enlargement of the spleen coincided with peak parasitemia in the absence of other gross lesions. Histopathology revealed lymphoid tissue hyperplasia and focal myocarditis. A canary that became blind and died of a systemic disease was examined (ARLT received slides for review from Gene Hubbard, VMD, Southwest Foundation for Biomedical Research, San Antonio, Texas): Heart and skeletal muscles showed typical inflammatory infiltrates and target cell lysis. The eyeball showed severe inflammation, with nests of dividing kinetoplastids in ciliary muscles. Pathology of the disease, caused by a natural trypanosome infection in birds, was similar to that of mammals infected with T. cruzi. Marsupialia The Metatheria (Marsupialia: Didelphidae) and Eu-theria (Edentata: Dasypodidae; Rodentia: Muridae) are considered the earliest mammals to become involved in the enzootic cycle of T. cruzi infection. Didelphidae and Dasypodidae, opossums and armadillos spp., respectively, are major sylvatic reservoirs (Legey et al. 1999, Yeo et al. 2005) The eco-epidemiology of enzootic Chagas disease in North America is dependent largely on the relationships of triatomine vectors with opossums and armadillos (Yaeger 1988, Karsten et al. 1992, Pung et al. 1995). T. cruzi infections among opossums ranged from 37.5% (Barr et al. 1991) to 57.1% (Ruiz-Pina and Cruz-Reyes 2002). Wild marsupials have been subjected to karyotyping and parasitological and pathological examinations (Teixeira et al. 2001). The karyotype confirmed the animals to be D. marsupialis. Nine out of the 12 marsupials had protozoan flagellates that were isolated by xenodiagnosis and/or hemoculture. The metacyclic forms recovered by xenodiagnosis were then inoculated in weanling mice. Two weeks after injection, trypomastigotes morphologically indistinguishable from T. cruzi were detected in murine blood. Phenotypic and genotypic molecular characterizations showed that these isolates were indeed T. cruzi, with a profile matching the Berenice stock, a laboratory standard for virulent T. cruzi. The pathology seen in heart sections from these naturally-infected marsupials showed myocarditis, characterized by mononuclear cell infiltrates and target cell lysis. Furthermore, inflammatory infiltrates were seen in the heart, skeletal muscle and in smooth muscle of the esophagus, and small and large intestines. Histopathological study of representative tissue sections taken from three opossums free of parasite showed an absence of tissue lesions (Teixeira et al. 2001). Araujo Carreira et al. (1996) found T. cruzi nests in the scent glands, hearts, and digestive tracts of 10 naturally-infected D. marsupialis. An inflammatory infiltrate of moderate to severe intensity was present in smooth and striated skeletal and heart muscles. Despite the presence of tissue lesions in wild T. cruzi-infected marsupials, armadillos, and rodents, some researchers have proposed that these animals "learned to live in harmony" with T. cruzi and, therefore they do not display apparent disease (Legey et al. 1999). No long-term study has shown ratios of morbidity and mortality or the relative lifespans of wild mammalian reservoirs in the presence or absence of T. cruzi infections. Rodentia Naturally T. cruzi-infected rodents (Rodentia: Echi-myidae; Rodentia: Cricetidae; and Rodentia: Muridae) have been captured in various ecosystems on the American continents. In one study, the prevalence of parasite infection reached 9.1% of captured wild rodents (Raccurt 1996). Calomys callosus (Rodentia: Cricetidae) are resistant to T. cruzi, surviving inocula that normally kill mice (Borges et al. 1992). The histopathological analysis of sections from T. cruzi-infected rodents showed parasitism of liver cells and of striated muscles. Inflammatory infiltrates in heart and skeletal muscles were moderate or absent (Borges et al. 1983). Resistance to chronic infection appeared to correlate with interferon gamma serum levels and H2O2 release by peritoneal macrophages. C. callosus may have developed immune mechanisms for survival and thus acts as a reservoir (Borges et al. 1995). The interaction of T. cruzi with the caviomorph rodent Trichomys apereoides (Rodentia: Echimyidae) revealed features suggestive of an ancient adaptation to T. cruzi infection. Chronic infection in T. apereoides remained pathologically cryptic for 5 months, despite persistence of infection (Herrera et al. 2004). Naturally occurring T. cruzi infection of sylvatic Rattus rattus and Rattus novergicus (Rodentia: Muridae) have been described (Herrera & Urdaneta-Morales 1997). Some T. cruzi-infected wild rats showed parasitemia and numerous nests of amastigotes in striated skeletal and cardiac muscle, and in smooth muscle of the digestive tract. In addition to showing myocarditis and myositis, inflammatory infiltrates could be seen occasionally in the proximity of amastigote nests. Over 9% of offspring showed transplacental T. cruzi infection in infected rats (Moreno et al. 2003). Several laboratory rat strains have been experimentally-infected with different T. cruzi stocks to assess their ability to produce myocarditis, histopathologic alterations in organs of the digestive system, and autonomic nervous systems denervation (Camargos et al. 2000). Extensive lesions in the heart of acutely-infected rats, accompanied by destruction of noradrenergic nerve terminals, were complement-independent (Machado et al. 1994). Chronic infection of rat revealed intra-cardiac destructive ganglionitis; the inventory of ED1 and ED2 macrophages and immune-competent cells infiltrating the ganglion was consistent with the fact that abundance of antigen presenting cells correlates with permeability of the blood-brain barrier and tissue lesions. Within the brain, astrocytes (Fig. 4A, B) are target cells for parasite proliferation (Da Mata et al. 2000). Peri-ganglionitis and ganglionitis were seen in 62.5% of the para-vertebral, sympathetic cervical ganglion from infected rats (Camargos & Machado 1988). Qualitative and quantitative morphology analyses suggest that infection of Wistar rats causes myelin damage and axonal swelling of myelinated fibers of the vagus nerve (Fazan & Lachat 1997). Among the Class Rodentia, small rodents in the Family Muridae are the most utilized laboratory animals for studies aimed at unraveling features of immunology and associated pathology in Chagas disease. The albino Swiss mouse (Mus musculus) is by far the most common animal in the study of parasite-host relationships in the course of experimental T. cruzi infection. Genetic control of immune responses in mice has been studied extensively, and this knowledge is useful for understanding the mechanisms of resistance and of susceptibility to T. cruzi. Existing knock-out mouse strains are important tools for determining the role of a selected gene in regulation of acquired immune responses in the course of infection (Araujo-Jorge 2000). Features of Chagas disease pathology have been unraveled in the laboratory mouse model. Briefly, myocarditis, myositis, sympathetic and parasympathetic ganglionitis, and central nervous system inflammatory infiltrates have been reported (Rossi Destetti 1995, Waghabi et al. 2002). In lesions at target tissues, the majority of mononuclear cells in inflammatory infiltrates bear surface markers characterized as CD8+ cells (Leavey & Tarleton 2003). Both CD4+ and CD8+ T cells participate in this process (Fuenmayor et al. 2005). Aberrant T-cell responses in Chagas disease are required to initiate immune responses that damage the heart (DosReis et al. 2005). Specifically, lesions of the autonomic sympathetic and parasympathetic nervous systems have been studied (Tafuri et al. 1971, 1979). At the level of optic and electron microscopy, Schwann and glial cells of nerve structures were found parasitized in the course of severe acute infections, while neurons were spared. The ultrastructural studies showed typical peri-ganglionitis, ganglionitis, and neuronolyses that were carried out by inflammatory mononuclear cell infiltrates (Tafuri 1971). Secretion of a hypothetical neurotoxin by T. cruzi that would kill neurons (Koeberle 1963) was discarded in acutely super-infected animals; mice receiving high doses of the immunosup-pressor hydrocortisone showed increasing numbers of parasitic forms of T. cruzi in Schwann and glial cells, but neurons were spared. In the absence of immunosuppression, acutely infected mice showed intense perigan-glionitis and ganglionitis, resulting in neuronolysis by inflammatory cells in infiltrates (Andrade & Andrade 1996). The advantage of using numerous isogeneic mouse lineages to reproduce features of human Chagas disease has been in jeopardy by a lack of definition of stable biochemical and physiological markers to characterize T. cruzi isolates and stocks kept in the laboratory (reviewed in Teixeira 1987). Furthermore, the events of genetic exchange in T. cruzi (Gaunt et al. 2003) underscore the instability of genetic features on the part of the parasite; reproducibility of some specific features of the infection could not be achieved, despite the use of isogenic mice. These features should not be considered as a caveat for using mice in experimental studies of Chagas disease, because in the wild, highly polymorphic parasite populations infect out-bred mammalian hosts and, therefore diversity is expected to be the common denominator of disease presentation. Nevertheless, the main pattern of T. cruzi infection in the laboratory mouse, although showing some variability, is characterized by a fulminating acute phase in which most infected animals, if not all, die within a few weeks of parasite inoculation. The infected mice attain high parasitemias and amastigotes can be easily found by histological exam of tissue sections. Although tissue parasitism can be intense, affected tissues may not show inflammatory infiltrates and target cell destruction. The highly virulent archetype Berenice and Tulahuen T. cruzi stocks, producing intense parasitism of the heart (Fig. 5A), skeletal and smooth muscles, and of the mononuclear phagocytic system, respectively, kill infected mice in two to three weeks. The cause of death in T. cruzi-infected mice is associated with high parasitemias and necrosis of the spleen. A variable percentage of acutely-infected animals may survive the acute phase, and then enter the chronic stage (Fig. 5B). Some consider mouse a suitable model for chronic Chagas disease (Marinho et al. 2004). The usefulness of this host cannot be underestimated; unquestionably, the mouse is suitable for the initial pre-clinical trials to determine the anti-trypanosomal activity and toxicity of candidate chemotherapeutic agents (Teixeira et al. 1994a,b). Lagomorpha The usefulness of the rabbit (Lagomorpha: Leporidae) has been recognized from the earliest experimental studies on Chagas disease (Chagas 1909). The rabbit (Ory-ctolagus cuniculus), inhabiting burrows in the ground or among rocks in the wild, can co-habit with triatomines, and is an important reservoir host in the cycle of T. cruzi transmission in some regions of the South American continent where they are domesticated. Rabbit scarcely had been used as a laboratory animal in experimental studies of Chagas disease, probably because animals are expensive to maintain in individual cages, and the lifespan is triple that of mouse. However, rabbits are highly resistant to T. cruzi infection and usually do not die in the acute phase, but later (20 ± 8 months) of chronic Chagas disease (Teixeira et al. 1975, 1983, Teixeira 1986); this advantageous feature of the rabbit has been recognized (Figueiredo et al. 1986, Silva et al. 1996). In one study, 34, one-month-old, out-bred New Zealand white rabbits received T. cruzi infections either intradermally, intravenously, or by drop instillation in the eye conjunctiva (Teixeira et al. 1983). Regardless of the route of infection the rabbits showed patent parasitemia by xenodiagnoses up to the 4th month post-infection. Thereafter, the xenodiagnoses were negative. Typical chagoma signs developed in two rabbits one week after receiving parasite inoculation in the skin, however the acute phase of the infection ran asymptomatically. In absence of direct demonstration of the parasite, the persisting cryptic infections were detected by serologic tests and delayed-type skin reactions to T. cruzi antigens. However, ECG alterations consistent with enlargement and overload of cardiac chambers, alterations of ventricular repolarization, S-T changes and bundle branch blocks were often recorded in the chronic phase. The pathological manifestations of these ECG alterations were confirmed at autopsy of experimental rabbits that died of Chagas disease (Fig. 5C). Congestive heart failure and pulmonary throm-boemboli related to chronic myocarditis were frequent causes of death. Megacolon was seen in two rabbits. The relatively limited duration of detectable parasitemia, the lack of correlation between parasitemia and severity of pathological manifestation, and the fact that rabbits showed histopathological evidence of myocarditis, myositis, ganglionitis, destructive inflammatory lesions characterized by mononuclear infiltrates and target cell lysis (Fig. 5D) are notable (Teixeira et al. 1983). Additionally, changes similar to those described in infected humans were produced in inbred III/J rabbits. Inflammatory infiltrates invaded the atria-ventricular node of the heart conducting system, where effectors cells adhered to the target myofibers. ECG alterations were recorded in chagasic rabbits, and increased cardiac silhouettes could be demonstrated in X-rays in the chronic phase (Teixeira 1986). Direct evidence of the cytotoxicity of the effector's immune lymnphocytes to the isogeneic target heart cells was provided by in vitro experiments: 73.5% of the beating target heart cell colonies ceased pulsating after a 2-h incubation with effector cells. This cell-mediated cytotoxicity bears implications for the physiopathology of arrhythmias and sudden death frequently seen in Chagas patients (Teixeira et al. 1983). Then I would still have this consolation - my joy in unrelenting pain - that I had not denied the words… The availability of the rabbit model for human disease prompted evaluation of treatment of experimental Chagas animals with anti-trypanosomal nitro-derivatives. A dose of 8 mg/kg/day for 60 days of nitroderivatives was intraperitoneally inoculated in infected and uninfected rabbits. Chronic infection was accompanied by the finding of myocarditis in every Chagas heart, regardless of treatment (Fig. 5E). PCR assays with T. cruzi-specific and nDNA-nested sets of primers yielded amplification products from Chagas rabbit DNA templates, regardless of treatment (Lauria-Pires et al. 2001). Thus, treatment of infected rabbits with nitroderivatives neither improved the Chagas heart lesions nor prolonged survival of treated animals (Teixeira et al. 1990a). Treated animals died in a time span comparable to infected, untreated rabbits. Alarmingly, malignant lymphomas (Fig. 5F, G) were seen in 33.3% of nifurtimox treated rabbits, and in 38.4% of benznidazole-treated rabbits (Teixeira et al. 1990a). In addition, interstitial fibrous thickening of the testes and scarcity of germinal cells in the seminiferous tubes of a benznidazole-treated rabbit was noted. Infected rabbits survived 765 ± 619 days post-infection whereas nifurtimox and benznidazol treated-infected rabbits survived 693 ± 434 and 552 ± 714 days, respectively (Teixeira et al. 1990b). These survival ratios were not statistically different from nifurtimox and benznidazol treated controls. All these survival ratios are significantly different from that (1496 ± 353 days) of control, untreated rabbits (p < 0.05). The myocarditis in treated rabbits was as intense as in the non-treated infected group. Myocarditis ranged in severity from focal to diffuse, with an even distribution among individual rabbits in each group. The survival of treated rabbits may have been affected by both myocarditis and the appearance of lymphomas in one-third of treated rabbits (Teixeira et al. 1990c). Infected as well as uninfected rabbits receiving nitroderivatives both developed malignant non-Hodgkin's lymphomas and died (Teixeira et al. 1990b,c, 1994a,b). Nitroderivative chronic toxicity should be measured in an epidemiologic scale, because nifurtimox and benznidazole resulted in lymphomas and atrophy of the testes. Carnivora Dogs (Carnivora: Canidae) have been recognized as important animal hosts participating in the peridomicile lifecycle of T. cruzi. Dog (Canis domesticus) mortality in natural ecotopes is substantial with high levels of transmission where dogs eat contaminated bugs. This clinical veterinary problem is recognized in several regions of the American continents. Naturally infected dogs have been identified in Texas, Louisiana, and Oklahoma (Bradley et al. 2000, Beard et al. 2003). In Cuesta Rica, it was shown that 27.7% of the dogs from five rural villages had T. cruzi-specific serum antibodies. Positive dogs subjected to X-ray and ECG revealed cardiomegaly and ECG alterations consistent with Chagas heart disease (Montenegro et al. 2002). T. cruzi infection of dogs as an experimental animal model of Chagas disease has been reviewed (Andrade et al. 1997). The scarcity of publications in this animal model is due to the long lifespan (15 years) and high costs of housing and feeding. Inoculated pups undergo a severe course of infection and usually die of acute Chagas disease. ECG alterations recorded two to four weeks post-infection consist of T waves and S-T abnormalities. Histopathological exams reveal mononuclear cell infiltrates with immune-effectors cell adherence to the membranes of target heart fibers, carrying out lyses and subsequent degeneration of parasite-free heart cells (Andrade at al. 1994). Immune effector cells likely play a major role in the pathogenesis of myofiber damage and microangiopathy in acute disease. Myocytes and parasitism of skeletal and smooth muscles, and multifocal encephalitis were recorded (Barr et al. 1991). Dogs that survive acute infection usually become asymptomatic. Histopathological exams reveal small, focal inflammatory infiltrates in the heart (Andrade et al. 1997), and the scarcity of lesions in the indeterminate phase explains the paucity of clinically-characterized chronic Chagas heart disease. In full-blown chronic heart disease the lesions were similar to those described in human. Inflammatory infiltrates in the atria-ventricular node of the conduction system of the heart are associated with lesions that correlate with ECG alterations (de Lana et al. 1992). Alterations related to Chagas disease have been identified independent of the number of superinfections imposed on experimental animals, consisting of focal, discrete myocarditis compatible with the indeterminate form of disease (Machado et al. 1994). Inflammatory infiltrates correlate with intracardiac ganglionitis and sympathetic neuron depopulation in chronically-infected dogs (Ma-chado et al. 1998). An interesting feature of T. cruzi infection in dog is the absence of mega syndromes (Machado et al. 2001). Primata The small Platyrrhini monkeys of the New World (Anthropoidea: Cercopitechoidae) are naturally-infected with T. cruzi and, thus, they play an important role in the epizootiology of the sylvatic cycle. Among 148 marmosets (Saguinus geoffroyi) captured in the Panama Canal Zone, 40% harbored T. cruzi (Sousa & Dawson 1976). Marmosets (Primate: Callitrichidae) inhabiting the Atlantic coastal rainforest have been found infected by T. cruzi zymodemes I and II (Lisboa et al. 2004a,b). The New World primates (Callitrix penicilata, Cebus apella, and Saimiri sciureus) are used as laboratory animals for T. cruzi infection. These primates are referred to as passive reservoirs (Bonecini-Almeida et al. 1990), however one-third of infected primates examined one year post-infection showed enlargement of the heart with effacement of the apex of the left ventricle (Rosner et al. 1989). The Old World Catarrhini primates serve as models for Chagas disease with experimental infection of Rhesus monkeys (Macaca mullata). Chagomas can be elicited at the site of inoculation in Rhesus monkeys; minor and transient ECG alterations were recorded in T. cruzi-infected monkeys, which are suggestive of chronic chagasic cardiomyopathy (Carvalho et al. 2003). T. cruzi infection and Chagas disease were detected in reared baboons (Papio hamadryas) at the Southwest Foundation for Biomedical Research, San Antonio, Texas. The colony originated with founder baboons imported from Saudi Arabia and were kept in large open-air pens. Serological data detected T. cruzi infections in 9.4% of 2-to-3-year-olds, 14% of 7-to-10-year-olds, and 22.5% of baboons that are 15-years-old or more. The primary vector for transmission is presumed to be reduviid bugs occasionally seen at night at the outdoor cages (Arganaraz et al. 2001). ECG and echocardiographic recordings revealed chagasic heart disease in 24% of the baboons with naturally acquired infections (Zabalgoitia et al. 2003). Flagellates recovered by hemoculture from three chagasic baboons were genotyped by in situ hybridization, showing traits of virulent T. cruzi (Arganaraz et al. 2001, Teixeira et al. 2001). The gross pathology of 12 chagasic baboons subjected to post-mortem exams revealed heart enlargement in five that died of chronic heart disease; megacolon and megaesophagus were also seen. In the acute cases the myocardial histopathological lesions consisted of parasite-free lysis of heart cells, with parasite nests seen in nearby tissue cells (Figs. 5H, I, J). The hearts of baboons that died of chronic disease showed severe myocarditis with inflammatory mononuclear cell infiltrates and target heart cell lyses. Amastigote nests were observed in host cells in regions of the myocardium free of inflammation. However, parasite nDNA remnants could not be detected by in situ hybridization in the region of the myocardium destroyed by immune system mononuclear cells (Fig. 5K, L). The central nervous system showed typical nodular lesions in the brain and mononuclear cells infiltrating the meninges. The parasympathetic ganglion in the esophagus and colon revealed typical features of neuritis, ganglionitis (Fig. 5M, N, O) and neuronal cell lysis, in the absence of parasite in target cells (EB Sousa, AC Ramos, GB Hubbard, ER Argañaraz, JL Vandeberg, and ARLT, unpublished data). The minimal rejection unit: a common denominator of pathology in Chagas disease Comparative pathology of Chagas disease in animal hosts from five Orders reveals common features that accompany T. cruzi infection. Persistence of the parasite is confirmed by direct demonstration or by indirect serologic and molecular markers of cryptic infection in each animal host. In infected mammalian hosts, however, histopathological lesions do not correlate with tissue parasitism; in wild reservoirs (marsupialia, rodentia, and primate) lesions appear to be less severe than those observed in man or some domesticated mammals. By contrast, acute infections in man and some laboratory animals are usually unperceived, and two-thirds do not die of Chagas disease. Pathologic lesions in the acute phase differ from those of chronic phase; only in the former are parasite nests easily seen. In chronic cryptic infection microscopic demonstration of the parasite in host tissues can be achieved in 10-20% of chagasics. A relevant feature of Chagas disease pathology is, thus, a lack of physical proximity between parasite nests and destructive inflammatory tissue lesions. Moreover, careful microscopic analysis of tissue lesions characterizes a relevant common denominator of pathology: the destruction of parasite-free target host cells by immune system mononuclear cell infiltrates. A unique pathological feature seen in every chagasic individual is inflammatory mononuclear cell infiltrates and lysis of target cells. This minimal rejection unit can be used to define pathology accordingly to the target tissue involved. In this respect, target units could affect either striated or smooth muscles, or neurons of the sympathetic and parasympathetic nervous systems. Although each of these pathologic lesions is detected in every chagasic, we favor the hallmark heart lesions associated with 94.5% of deaths. Therefore, a typical `minimal rejection unit' is defined here as a destructive lesion imposed upon parasite-free target heart cells by surrounding mononuclear cells of the host immune system (Fig. 6). We next focus on the importance of the minimal rejection unit as well as parasite persistence for an understanding of the pathogenesis of Chagas disease. Pathogenesis of Chagas disease Questions related to the mechanism by which tissue lesions are formed in the course of T. cruzi infection have long been a matter of contentious debate. Although numerous reviews have proposed a plethora of brilliant hypotheses (Levin 1996, Kierszenbaum 1999), the origin of the pathological lesions of Chagas disease remains open to investigation. Parasite persistence The earliest hypothesis addressing pathogenesis of Chagas disease stems directly from identification of the parasite in tissues. A precise description of the pathology of acute disease led to the idea that the disease stems from microbial infection. Accordingly, mechanical rupture of parasite nests and degradation of affected tissues could drive chronic inflammation. Nevertheless, the main difficulty in establishing a direct relationship between T. cruzi infection and endemic chronic disease was the absence of parasite nests in pathological sections of tissues from 80% of deceased patients. This difficulty has been resolved with specific immunological tests for indirect identification of parasite antigens and of sensitive genetic markers showing persistence of parasite kDNA in tissues of Chagas patients (Zhang & Tarleton 1999). However, only nDNA detection is indicative of an active T. cruzi infection (Braga et al. 2000, Lauria-Pires et al. 2000) Key questions remained unanswered: (i) why are acute infections usually clinically unperceived before subsiding spontaneously? (ii) Is tissue damage seen in cryptic infections for all patients, or only some patients? (iii) Why do patients showing cryptic chronic chagasic infection have high mortality ratios? Partial answers have been sought in field studies. Among 190 seropositive chagasics, 134 showed negative parasitemias, and 56 had parasitemias detected by xenodiagnoses. Progressive Chagas heart disease was verified, respectively, in 30% with positive xenodiagnosis (high parasitemia) and in 28.8% showing negative xenodiagnoses (Castro et al. 2005). Thus, the heart lesions cannot be associated with the severeness of parasitemias in chronic Chagas patients. Unifying neurogenic A neurogenic hypothesis was once in vogue to explain Chagas pathogenesis (Koeberle 1970). A modified neurogenic hypothesis has been presented aimed at unifying the bulk of theories to explain the chronic lesions of Chagas heart disease (Dávila et al. 2004). Some believe that impairment of the parasympathetic nervous system and permanent activation of the sympathetic nervous system and of other neuro-hormonal circuits explains the lesions (Bestetti et al. 1995, Dávila et al. 2002). This hypothesis (Dávila et al. 1998, 2005) is based on data showing that chronic patients with parasite persistence but without myocardial damage do not have cardiac parasympathetic impairment or neuro-hormonal activation. Abnormalities of the autonomic nervous system and autoimmune reaction initiate and perpetuate the vicious cycle of catecholamine cardiotoxicity, myocytolysis, and heart failure. However, the unifying neurogenic hypothesis does not address the origin of Chagas disease pathogenesis. Autoimmunity In the last three decades autoimmunity has been considered an important pathogenic mechanism receiving substantial experimental support and attention (Girones & Fresno 2003, Leon & Engman 2003). The key pathological feature of the parasite-free target cell destruction carried out by inflammatory mononuclear cell infiltrates was explored, emulating in vivo and in vitro experimental conditions. With this aim, an accelerated rejection of allogenic heart cells by immune lymphocytes from chronically infected rabbits was described (Santos-Buch & Teixeira 1974). Target embryo heart cells were readily destroyed by immune lymphocytes within an hour, whereas lymphocytes from controls were not. This observation prompted the autoimmune hypothesis (Teixeira 1975). Mechanisms of autoimmune triggering are a matter of debate, stimulating discussion on the role played by T. cruzi antigens, cross-reacting epitopes or by molecular mimicry . We direct the reader to previous reviews (Leon & Engman 2003, Tarleton 2003). Roles played by the persisting parasite and by autoimmunity may be essential in Chagas pathogenesis. To clarify a role for autoimmunity it is important to address cause and effect. According to Kock's postulates an infectious agent must be consistently isolated from an infected host at every stage of disease. Microbes isolated from the diseased host must grow in culture or laboratory animal, from which it could then be recovered. Next, upon inoculation in a suitable host, the recovered microbe should be able to transmit the disease to a healthy individual, showing features seen in the diseased host from which it was isolated originally. Finally, the microbe must be re-isolated from the diseased animal and matched to the original microorganism in pure culture. Kock's postulates cannot be fulfilled in a great majority of pathologies in the absence of a demonstrable infectious agent. For Chagas disease, however, potential contamination of immune cells with live T. cruzi prevents fulfillment of Kock's postulates. In this respect, to determine a role for autoimmunity in Chagas disease requires production of lesions by passive transfer of the effector's immune cells. Stemming from the development of progressive Chagas heart disease in infected rabbits treated with anti-trypanosomal nitroderivatives (Teixeira et al. 1990b,c) the following question became inevitable: What could be sustaining active destruction of heart cells in treated rabbits? We demonstrated that a high rate of genetic transfer occurs from the parasite to the host genome and that resulting mutation could explain persistence of autoimmune-driven lesions in Chagas disease (Nitz et al. 2004). Then, we hypothesized that Chagas autoimmunity could be triggered by phenotypic changes induced by parasite DNA retained in the host genome. Horizontal transfer of minicircle kDNA from T. cruzi to the host genome An answer to the origin of autoimmunity in Chagas disease was sought in genomic DNA from the human macrophage line U937 that had been infected (ratio 1:5) with trypomastigoes of T. cruzi (Simões-Barbosa et al. 2006). All samples assayed harbored parasite DNA as judged by NAT molecular markers to T. cruzi. Interestingly the kDNA integrated in copies of LINE-1 (GenBank AF002199 to AF002203) showing similarity with the human ORF2 transcribing the human reverse transcriptase. Horizontal transfer of minicircles was observed in Chagas patients (GenBank AY490906, AY490905, AY490902). Active transfer of minicircles was observed in DNA from chronically infected hosts. In the human cell the kDNA integrates more frequently at the chromosomes 3, 6, and 11 (Teixeira et al. 1994a,b, Nitz et al. 2004). Also, truncated minicircle sequences were found integrated, flanked by rearranged host DNA from chagasic baboon receptive to T. cruzi (GeneBank DQ241812). Next we demonstrated that T. cruzi kDNA, specifically minicircle sequences, was transferred to Chagas rabbit (Nitz et al. 2004). Digestion of rabbit DNA from blood, heart, skeletal muscle, liver, intestine, and kidney were hybridized with a constant region minicircle probe. Fig. 7A illustrates the altered configuration of the band pattern of kDNA integrated in the heart, intestine and skeletal muscle, distinct from minicircle unit size (1.4 kb), seen in parasite DNA alone. These tissue samples showed no hybridization with other T. cruzi-specific nDNA probes (cited in Nitz et al. 2004). Sequencing of the 2-kb band revealed rabbit DNA flanking the minicircle insertion (Fig. 7B). Minicircle integration occurred at sites of direct CACCAACC repeats within rabbit DNA (GenBank AF400668, AF399841, and AF415293). The flanking DNA showed homology with the LINE-1 clone LBNL-1125D4, and contained interspersed SINE repeats (Price et al. 1992). An ORF initiating in the host DNA and extending through the kDNA integration could generate a transcript encoding a chimeric r45-like antigen (GenBank AAR24603.1). In a specific case the insertion mutation was comprised of 27 truncated kDNA fragments of diverse size and structure, indicating that this genomic mutation was initiated by tandem insertions of seven full minicircle sequences, comprising a grand total of 10.8 kb of kDNA (Nitz et al. 2004). Also, multiple (four or more) mutation events in the genome of Chagas patients suggest that horizontal transfer of minicircles could be part and parcel, and indeed a direct cause, of genome growth and evolution. Single kDNA integrations occurring at multiple sites could explain the variability of clinical manifestations. Accumulation of kDNA integration-induced mutations may be a force naturally driving pathology in Chagas disease. The vertebrate genome contains stretches of long and short repetitive sequences (SINE and LINE elements) that perpetuate themselves by vertical transmission within the host (Smit et al. 1995, Furano et al. 2004). The human genome contains 535 LINEs belonging to the family Ta and 415 in the subfamily Tn. Thirty-nine elements of the Ta family and 22 of the subfamily Tn show the standard 6.4-kb sequence with a 5' promoter followed by two ORFs and a 3' UTR with polyadenylation signal and a poly-A tail, features of active LINE-1 retrotransposons (Feng et al. 1996). These elements are recognized progenitors of mutagenic insertions, because they possess endogenous machinery including transposase, a DNA polymerase I and reverse transcriptase combination, for mobilization of DNA sequences from within the genome, thus generating exon rearrangement (Gilbert et al. 2002, Symer et al. 2002). The 5' promoter initiates transcription of LINE-1, which is generally confined to germ line cells (Trelogan & Martin 1995, International Human Genome Sequencing Consortium 2001), and retrotransposition of LINE-1 in somatic cells has been correlated with genetic diseases (Kazazian & Moran 1998, Ostertag & Kazazian 2001). Integrated kDNA heritability and pathogenesis If these observations are validated, we have created a powerful new research tool for the understanding of Chagas disease (Nitz et al. 2004) Acute Chagas pathology served as an indicator for examining if T. cruzi infection could be established in the goniablasts of the testes and the germinal theca cells of the ovary (see section acute pathology). Although invasion of embryonic stems cells had not been demonstrated, we assayed parasite uptake by stem cells in vitro: embryo stem cells from a 2.5-day-old zygote actively engulfed trypomastigotes (Nitz et al. 2004). Amastigotes loaded the cytoplasm of rabbit embryonic cells, and similar kinetics was observed with infected chicken embryonic cells. The permissiveness of embryonic stem cells to T. cruzi suggested that differentiating cells in the genital crest, which appear at days 4-8.5 of gestation, could acquire integrated kDNA-induced mutations during early parasite invasion. Thus, embryonic cells became candidates for vertical or germline transfer of kDNA integration-associated mutations. Transplacental transmission of T. cruzi and subsequent kDNA integration was attempted experimentally in litters from chronically-infected Chagas rabbits. Four sexually mature does and two bucks infected with T. cruzi were crossbred during chronic infection. Three pregnancies resulted in 104 offspring. NAT assays on stillborn animals or surviving offspring of infected rabbits were performed for parasite nDNA and kDNA, revealing that 15 (14.4%) contained nDNA suggestive of active infection, and 24 (23%) contained kDNA only. Stillborn offspring yielded DNA from heart, skeletal muscle, liver, spleen, and large and small intestine, and each tissue type was positive for specific kDNA probes. In the case of a chronically-infected pregnant doe that delivered six offspring, five showed positive NAT for kDNA; one yielded positive NATs for kDNA and nDNA. Thus five presented vertical transfer of kDNA via the parental gametes, whereas only one received a transplacental living infection (Fig. 8A, B). Genomic DNAs of kDNA-positive offspring were subjected to 5' RACE, yielding six integration sites of minicircle fragments (GenBank AY488498 to AY488503). Typical histopathological lesions in muscle tissues, which were usually extensive in the peripheral nervous systems of T. cruzi-infected rabbit offspring, were similar to those lesions seen in chagasic rabbits and humans (see human pathology and lagomorpha sections). A living infection is necessary for kDNA integration. With the demonstration of vertical transfer of kDNA from chronically-infected rabbits to offspring, the high frequency with which parasite kDNA could be inherited by its host in a Mendelian fashion was evident, along with the presence of kDNA fragments in diverse tissue types (Fig. 9A, B). To eliminate persistent infection as a requirement for kDNA integrations in the host genome, experiments were conducted in chickens that are refractory to chronic T. cruzi infection. Thus, the permissiveness of chicken embryonic cells to experimental T. cruzi infection was shown, making dissociation of the kDNA integration event and pathology from the presence of the active infection possible. Minicircle kDNA integrated at the chicken chromosome 4 (GenBank AY237306). Use of the aves model provided the basis for parasite-free pathology in chicken that was indistinguishable from human Chagas pathology (Nitz et al. 2004). Introduced kDNA was present in 25% of chicks hatched from T. cruzi-inoculated eggs. Due to the refractory nature of the chicken to T. cruzi infection, the pathological lesions described in kDNA-positive chickens eliminate any role of parasite persistence. With establishment of germline kDNA integration in the chicken genome, vertical transfer of T. cruzi DNA to infection-free progeny was demonstrated. Interestingly, the birds from F0 and F1 progeny developed signs of widespread muscle weakness, with some individuals not strong enough to stand. Usually chickens showing this systemic disease died young: Striated heart and skeletal muscle, smooth muscles and the parasympathetic ganglia showed typical lesions seen in chagasic mammals (Fig. 10A, B, C, D). The parasite-free Chagas heart disease in chicken showed minimal rejection units as seen in human Chagas patients, characterized by mononuclear infiltrates and lyses of target heart cells (Fig. 11A, B, C, D). Thus, kDNA integration represents a likely cause for the autoimmune response and may be the key to understanding chronic Chagas disease manifestation (Nitz et al. 2004). Numerous instances of kDNA integration into genomes of vertebrates infected with T. cruzi have been documented. Mapping of kDNA insertion sites, including active mobilization via the LINE-1 transposition machinery (Symer et al. 2002), may correlate with the variable and delayed clinical manifestations. LINE sequences polluted the vertebrate genomes over 150 mya, prior to speciation of Homo sapiens. The vertebrate genome is filled (> 50%) with repeat sequences, including interspersed repeats derived from transposable elements (LINEs carrying SINEs on their back), and long genomic regions that duplicate in tandem, palindromic or dispersed fashion. These include duplicated segments, at which mispairing during recombination creates deletions responsible for genetic syndromes (Ostertag & Kazazian 2001). Both kDNA and host DNA are rearranged: the kDNA insertions and host flanking regions are subject to the consequences of deletion and rearrangement. Therefore, genomic DNA samples showing horizontal transfer of minicircles into the host vertebrate genome are useful laboratory tools for the calibration of molecular clocks. Complete genome sequencing from an archetype Chagas patient with deletions and recombination resulting from kDNA integrations would give a unique opportunity to advance research in this field. The chicken genome database has been completed and its annotation is available. There are over 200,000 copies of the repetitive retrotransposon CR-1 (equivalent to LINE-1 of mammals) in chicken, about twice the amount found in mammals. In addition approximately 10,000 SINE matches were found, similar to mammals (International Chicken Genome Sequencing Consortium 2004). Concluding remarks Any one whose disposition leads him to attach more weight to unexplained difficulties than to the explanation of a certain number of facts will certainly reject my theory.(Charles Darwin, The Origin of Species, 1859) Horizontal DNA transfer experiments were reproduced in mammals permissive to infection and in birds non-permissive to T. cruzi. The demonstration of kDNA minicircle integration into germ line cells of birds represents a clean biological system for showing horizontal gene transfer, as chickens are refractory to persistent infection. kDNA-positive chicks hatched from infected eggs demonstrate pure kDNA transfer that cannot stem from residual live parasite contamination. Further vertical transfer of T. cruzi DNA to the progeny of kDNA-positive birds was obtained by crossing. Some of these kDNA-positive birds present a generalized muscular weakness that evolves into death of the affected animal. ECG alterations typical of Chagas heart disease have been recorded in these birds. Interestingly, some kDNA positive hens and roosters present signs of heart insufficiency, such as cyanosis and shortness of breath. These animals show lesions typical of Chagas heart disease (Fig. 12A, B). The phenomenon of kDNA integration is critical for understanding chronic Chagas disease. In several animal models, pathogenesis of Chagas disease is revealed as a parasite-kDNA vector phenomenon eliciting confluent minimal rejection units, the common denominator of autoimmunity in Chagas disease. Typical pathological lesions in mammals were similar to those evident in birds. In considering this pathology, why do the immune effector cells change from physiological to pathological behavior? Clearly kDNA integration that takes place in early embryonic life can be perpetuated through the germ line: The mutated somatic host cells could induce phenotype modifications that trigger autoimmunity in Chagas disease. The kDNA and host DNA juxtaposition carries the potential to induce anti-self immune responses with the expression of novel chimeric proteins. In favor of this model is the demonstration of Chagas lesions in kDNA-mutated birds: kDNA-integration induced mutation in the course of Chagas disease is associated with the histopathology evident in host tissues. Future investigations will open new avenues to explore three non-mutually-exclusive hypotheses: (a) auto-reactive effector immunocyte heterogeneous populations, reactive to chimeric proteins formed by kDNA incorporated into the genome of host cells, can be identified in tissue lesions, isolated from blood or lymph nodes, and used in assays of rejection of syngenic heart grafts; (b) auto-reactive lymphocytes against target host cells present in the chronic inflammatory immune response could be induced directly by parasite antigens, identified in the tissue lesions, isolated from blood or lymph nodes, and used in pathology assays; (c) auto-reactive lymphocytes showing somatic mutation and phenotype changes could be identified in tissue lesions, isolated from blood or lymph nodes, and used in pathology assays. In hypothesis a, the immune effectors cells would react against modified (e.g., mutated) somatic target cells. In hypothesis b and c, the self-reactive forbidden clones would also destroy normal (e.g., non-mutated) target host cells. The identification, cloning, and amplification of such forbidden clones will generate genotype and phenotype data prior to obtaining passive transfer of autoimmunity. Dispersion of kDNA mutations in the population, representing a force towards evolutionary change, is expected. Along these lines, some level of change in gene frequencies will occur over generations. Normally this dispersal is achieved by sexual reproduction of interbreeding populations. In vertebrate hosts, kDNA-insertion mutation could play different functional roles ranging from advantageous to neutral. Genetic drift can rapidly fix the advantageous and neutral mutations that may associate emergence of adaptive characters over time, fixation being the prevailing mechanism of evolution at the molecular level. It is particularly important to consider that truly deleterious kDNA mutations have not been demonstrated in the reported investigations. By definition, true deleterious or purifying mutations preclude the reproductive saga leading to perpetuation of the species. In the case of Chagas disease, individuals often live a standard reproductive life, but their descendants may carry the molecular baggage of parental infection. Ongoing observations indicate that Chagas pathology kills a greater number of founder (FO) than F1 and F2 birds, and gradually faded in the third generation (FO > F1 > F2 > F3). These observations are consistent with our thoughts that autoimmunity in Chagas disease is a purely fortuitous sharing of negative selection, with biological effects that tend to be modulated naturally over time in due benefit to evolution of the species. A goal of future work is to demonstrate whether microbial infections triggering horizontal gene transfer and further vertical transmission to progeny can cause autoimmune rejection of target cells mediated by the host immune effector cells. Passive transfer of the lesions from a mutated syngenic donor to a healthy receptor will be required. The hope is that we can now unravel the pathogenesis of this reputably untreatable autoimmune disease. ACKNOWLEDGMENTS To Ana de Cássia Rosa, Márcia Peres, and Migues Ribeiro dos Santos for their excellent technical assistance, and to our colleagues Ana de Cassia Rosa, Ana Maria Barros, Augusto Simões-Barbosa, Enrique R Arganaraz, Clever Gomes, Jaime M Santana, Liana Lauria-Pires, Mariana Hecht, and Nadjar Nitz, from the Chagas Disease Multidisciplinary Research Laboratory, where some of these studies were carried out. REFERENCES
Copyright 2006 Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc06082t1.jpg] [oc06082f4.jpg] [oc06082f6.jpg] [oc06082f7.jpg] [oc06082f12.jpg] [oc06082f8.jpg] [oc06082f11.jpg] [oc06082f10.jpg] [oc06082f3.jpg] [oc06082f1.jpg] [oc06082f5.jpg] [oc06082f9.jpg] [oc06082t2.jpg] [oc06082f2.jpg] |
| |||||||||

{kind=link}
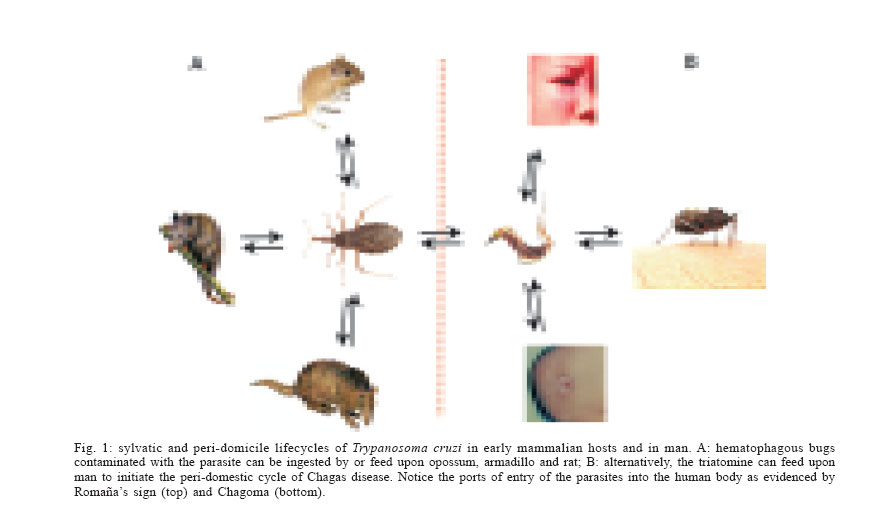{kind=link}
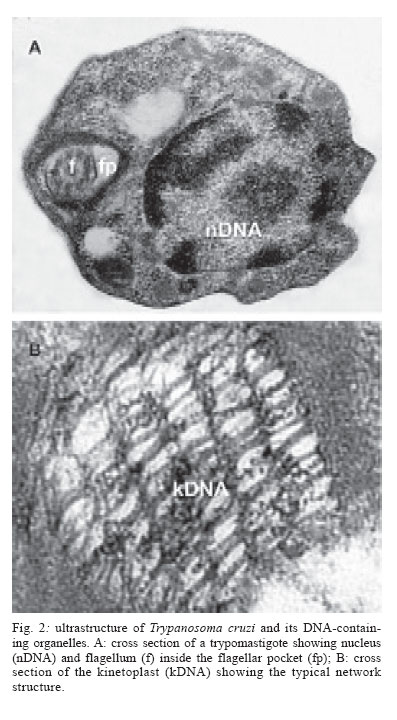{kind=link}
{kind=link}
{kind=link}
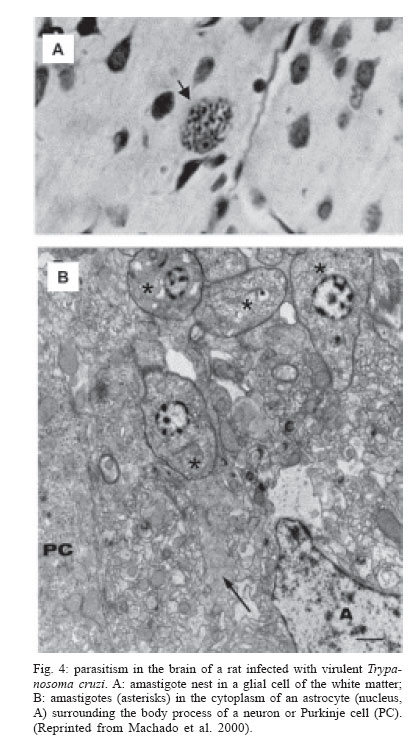{kind=link}
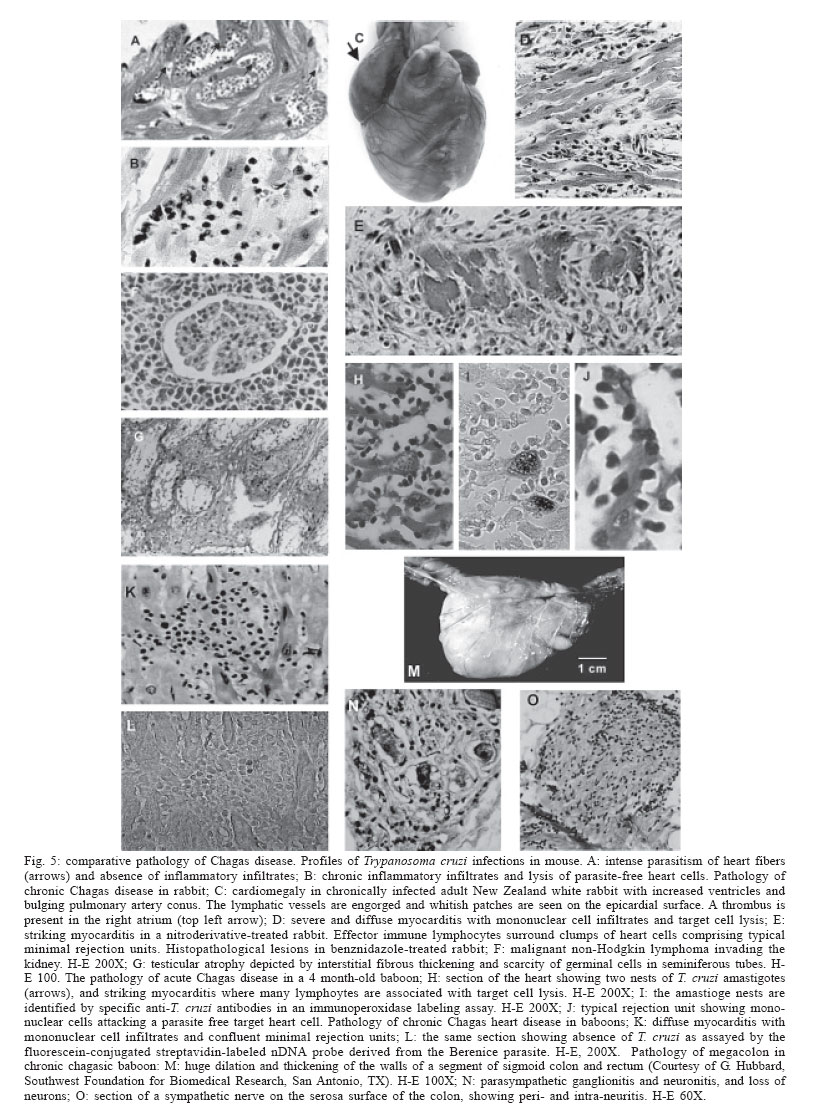{kind=link}
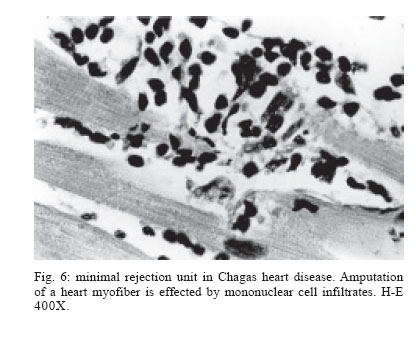{kind=link}
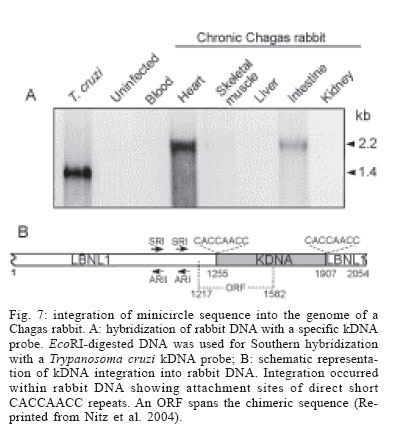{kind=link}
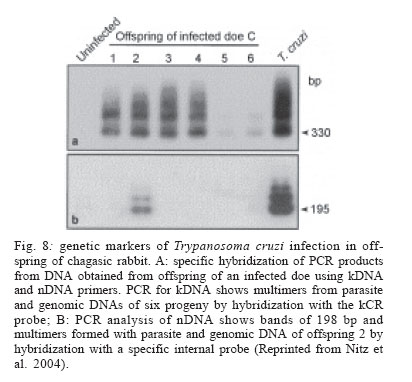{kind=link}
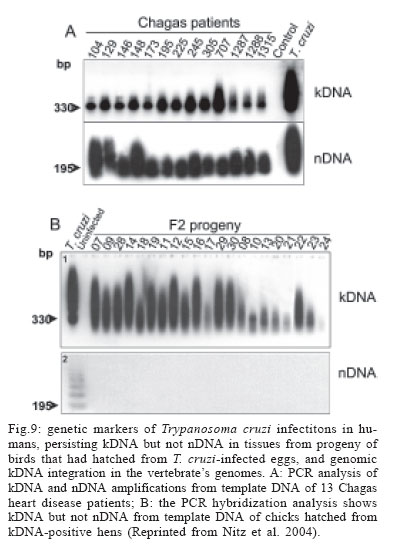{kind=link}
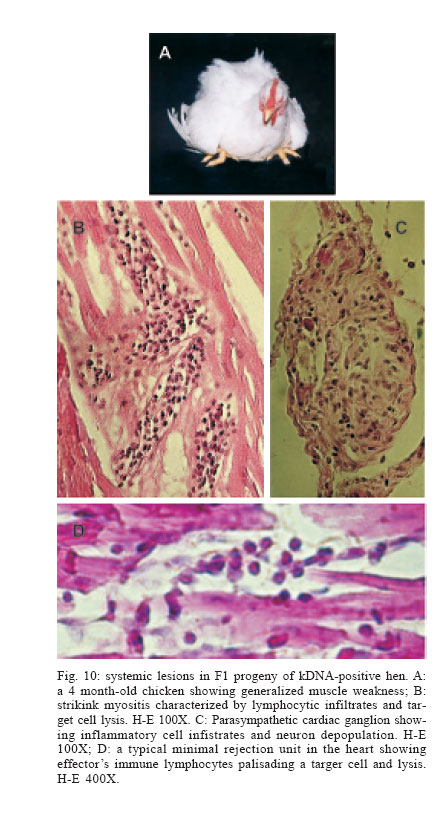{kind=link}
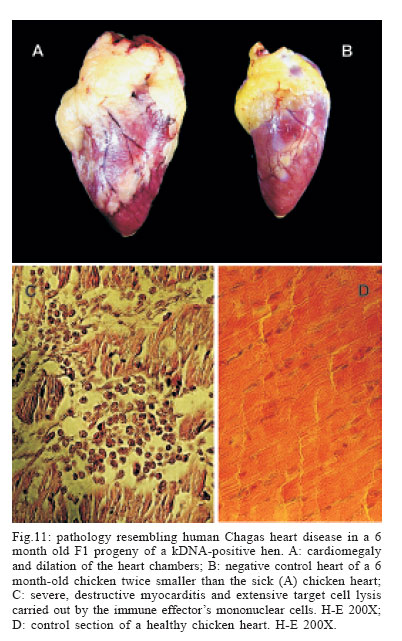{kind=link}
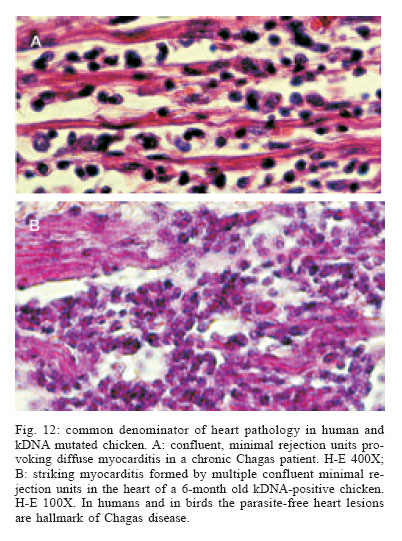{kind=link}