 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol. 101,Suppl. I, October ,2006, pp. 161-165 Production of full-length cDNA sequences by sequencing and analysis of expressed sequence tags from Schistosoma mansoni Alessandra C Faria-Campos, Fernanda S Moratelli, Isabella K Mendes, Paula L Ortolani, Guilherme C Oliveira*, Sérgio V A Campos**, J Miguel Ortega, Glória R Franco/+ Departamento de Bioquímica e Imunologia, Instituto
de Ciências Biológicas **Laboratório de Universalização
de Acesso a Internet, Departamento de Ciência da Computação,
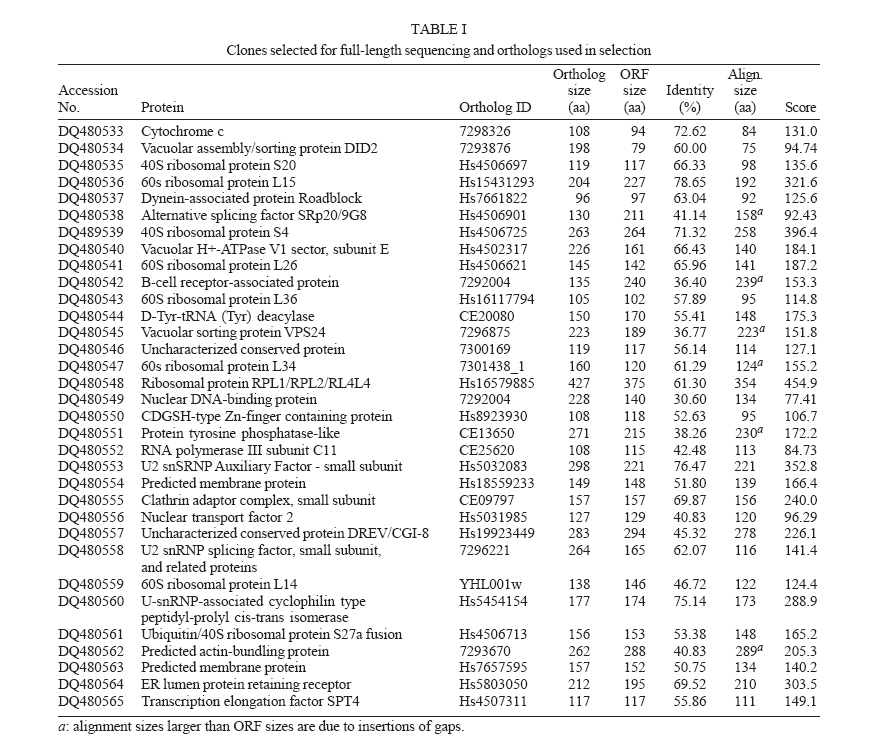
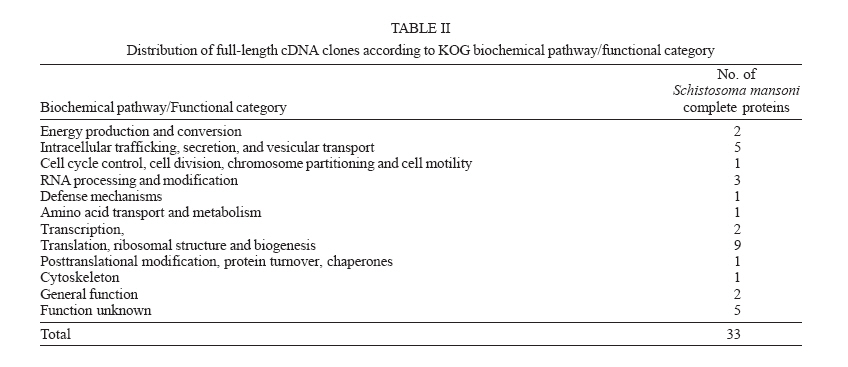
Instituto de Ciências Exatas, Universidade Federal de Minas Gerais, Financial support: CNPq, Fapemig Received 26 May 2006 Code Number: oc06184 The number of sequences generated by genome projects has increased exponentially, but gene characterization has not followed at the same rate. Sequencing and analysis of full-length cDNAs is an important step in gene characterization that has been used nowadays by several research groups. In this work, we have selected Schistosoma mansoni clones for full-length sequencing, using an algorithm that investigates the presence of the initial methionine in the parasite sequence based on the positions of alignment start between two sequences. BLAST searches to produce such alignments have been performed using parasite expressed sequence tags produced by Minas Gerais Genome Network against sequences from the database Eukaryotic Cluster of Orthologous Groups (KOG). This procedure has allowed the selection of clones representing 398 proteins which have not been deposited as S. mansoni complete CDS in any public database. Dedicated sequencing of 96 of such clones with reads from both 5' and 3' ends has been performed. These reads have been assembled using PHRAP, resulting in the production of 33 full-length sequences that represent novel S. mansoni proteins. These results shall contribute to construct a more complete view of the biology of this important parasite. Key words: expressed sequence tags - sequencing - Schistosoma - full-length cDNA While the number of sequences generated by genome projects has increased exponentially, this phenomenon has not been followed by gene characterization at the same rate (Saghatelian & Cravat 2005). Aiming to diminish that gap, several approaches have been used for gene discovery through searches on the genomic sequence or analysis of the transcriptome of the organisms (Okazaki et al. 2002). One such approach is gene discovery using expressed sequence tags (ESTs), short sequences produced by sequencing the ends of cDNA molecules, which represent a snapshot of the gene expression for a given organism at a certain time (Adams et al. 1991). However, a complete picture of the gene products of the organism can only be obtained when the full-length sequence of specific proteins is determined. For that, it is essential to select clones that potentially present the complete coding sequence, up to the initial methionine and proceed to sequencing and characterization of such clones (Das et al. 2001). The initial step in the characterization is to determine the completeness of the cDNAs from which the ESTs were generated, followed by the annotation using the biological description of sequences present in function-oriented databases. Several authors developed systems to attain this goal which use among other tools similarity searches, statistical information and genome mapping (Salamov et al. 1998, Nishikawa et al. 2000, Del Val et al. 2003, Furuno et al. 2003, Hotz-Wagenblatt et al. 2003). Schistosoma mansoni cDNAs have been selected in this work for full-length sequencing using an algorithm based on BLAST searches of parasite ESTs against sequences from the database Eukaryotic Cluster of Orthologous Groups (KOG) (Tatusov et al. 2003). The implementation of the algorithm uses SQL searches to predict the presence of the initial methionine in the parasite sequence, resulting in the selection of ESTs representing clones putatively containing the complete sequence. By this procedure we have been able to select 3988 ESTs representing 398 proteins which have not yet been deposited as S. mansoni complete CDS in any public database. Dedicated 5' and 3' end sequencing of 96 clones has been performed and reads have been assembled using PHRAP. As a final result, 33 full-length sequences have been produced which represent novel S. mansoni proteins. MATERIALS AND METHODS Selection of ESTs representing putative full-length clones - S. mansoni ESTs have been selected for full-length sequencing using the algorithm described by Nishikawa et al. (2000) with modifications. The completness of the clones represented by the ESTs was determined by comparison of these to sequence of proteins from the secondary database KOG using tblastn. The positions of start and end of alignment assigned by BLAST in both sequences were determined through SQL queries. When the length of the not-aligned 5'-terminal of the EST was longer than that of the beggining of the aligned protein multiplied by three, the EST has been considered to represent a clone that had the entire coding region and therefore has been selected for full-length sequencing (Fig. 1). A total of 63,960 ESTs produced by Minas Gerais Genome Network (RGMG) has been used in BLAST searches against 88,644 sequences from the database KOG. Start and end alignment position from BLAST results have been inserted into a MySQL database for processing, allowing clone selection through SQL queries. Sequencing and assembling - DNA has been prepared for sequencing as described by Brazilian National Genome Project Consortium (2003), using 400 hg of DNA. Sequencing reactions have been performed using the kit DYEnamic™ ET Dye Terminators according to instuctions provided by the manufacturer with the primers M13 reverse (5'-GGAAACAGCTATGACCATG-3') and foward (5'-GTTTTCCCAGTCACGAC-3') and run on MegaBACE™ 1000 (GE Healthcare). Six reads (three for each primer direction) have been generated for each clone ranging from 0.4 to 0.6 Kb. Base calling and sequence assembling have been performed using phred/phrap/consed (http://www.phrap.org) and a quality value cutoff phred = 20. Annotation and search of ORFs - Contig annotation has been performed using BLAST searches against KOG sequences and the tool Blast2GO (Conesa et al. 2005). Manual inspection of sequences has been performed after automatic annotation and ORFs have been located by Consed analysis. Complete sequences have been translated using RevTrans (Wernersson & Pedersen 2003) and ORFs aligned to their ortologs using Multialin (Corpet 1988) to confirm completeness. Results By running an implementation of the clone selection algorithm on EST sequences generated by RGMG consortium, a total of 3988 clones putatively having the full-length sequence has been selected. These clones represented 398 proteins which have not been deposited as S. mansoni complete CDS in any public database. From these, 96 clones have been re-sequenced and assembled. Assembly resulted in 33 contigs representing novel S. mansoni proteins with sizes ranging from 79 to 375 amino acids with identities in the alignments varying from 30.6% to 78.65% (Table I). These proteins belonged to 11 different KOG biochemical pathways/functional categories as seen in Table II: Energy production and conversion; Intracellular trafficking, secretion, and vesicular transport; Cell cycle control, cell division, chromosome partitioning and cell motility; RNA processing and modification; Defense mechanisms; Amino acid transport and metabolism; Transcription; Translation, ribosomal structure, and biogenesis; Posttranslational modification, protein turnover, chaperones, and Cytoskeleton. Eight proteins have not been classified regarding a biochemical pathway, belonging to the category of General Function or Function Unknown. Selected sequences have been translated and aligned to their orthologs, to verify completeness. Fig. 2 shows an alignment for one of the proteins as an example. Alignments for all proteins can be seen on supplementary material (www.icb.ufmg.br/~alessa/pesquisa/pesquisa.html). Complete sequences have been submitted to NCBI. Discussion Full-length cDNAs are extremely useful for determining the genomic structure of genes, especially when analyzed within the context of genomic sequence (Strausberg et al. 2002). Knowing the full-length sequence of a gene allows the prediction of the entire sequence of a protein which can be used for functional and evolutionary studies and for improving the knowledge of the biology of this species. However, the selection of specific cDNAs for full-length sequencing made without the aid of bioinformatics tools is very laborious and time-consuming since it involves the screening of a number of cDNA libraries of variable quality and/or direct strategies to process individual clones (Das et al. 2001). In this study we proposed a method that can be used to increase the availability of full-length sequences, and applied the algorithm to S. mansoni sequences. The selection of such clones by computer speeds up the investigation process and sequencing of such clones provides an approach to investigate S. mansoni sequences that have not been used by other groups yet. Many initiatives for the investigation of complete CDS in several organisms, including S. japonicum, are currently under way and this work now integrates these efforts (Strausberg et al. 1999, Collins 2002, Hu et al. 2003, Baross et al. 2004). The number of S. mansoni complete CDS sequences publicly available before this work (Feb/2006) was of 437 nucleotide sequences described as complete CDS in GenBank flat files and 1108 protein sequences having a suggested CDS pointed in the GenPept files. However, a great number of the CDS sequences available at the moment represent redundant entries in the database. The sequences obtained in this study are unique sequences representing proteins not previously described as complete and therefore representing an important contribution in functional gene characterization of this parasite. The proteins analyzed in our study indicate a high degree of conservation with the orthologs used in the selection with small variations in size and sequence (see alignments in supplementary material). This is also suggested by the values of identities in the alignments presented in Table I. We have not observed a high variation in any specific portion of the proteins studied, although previous studies suggested a higher degree variation in the amino terminal portion of eukaryotes proteins (Kisselev & Frolova 1995). We observed that the size of the proteins for which the full-length sequences have been obtained was relatively small, as should be expected given the characteristics of the procedure used for library construction for production of ESTs. In this study we have proposed a method that can be used to increase the availability of full-length sequences and used this method to study S. mansoni sequences. This represents an important contribution in the research on this parasite, for which the complete functional characterization of the genome is still under way. Acknowledgments To Minas Gerais Genome Network for providing ESTs for analysis REFERENCE
Copyright 2006 Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc06184f2.jpg] [oc06184t1.jpg] [oc06184f1.jpg] [oc06184t2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}