 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 102, No. 5, 2007, pp. 573-580 Genetic variability of Aedes aegypti in the Americas using a mitochondrial gene: evidence of multiple introductions José Eduardo Bracco/***/+, Margareth Lara Capurro*, Ricardo Lourenço-de-Oliveira**, Maria Anice Mureb Sallum*** Laboratório
de Bioquímica e Biologia Molecular de Vetores, Superintendência
de Controle de Endemias, São Paulo, SP, Brasil *Departamento
de Parasitologia, ICB, Universidade de São Paulo, São
Paulo, SP Brasil **Departamento de Entomologia, Instituto Oswaldo
Cruz-Fiocruz, Rio de Janeiro, RJ, Brasil ***Departamento de Epidemiologia,
Faculdade de Saúde Pública, Universidade de São
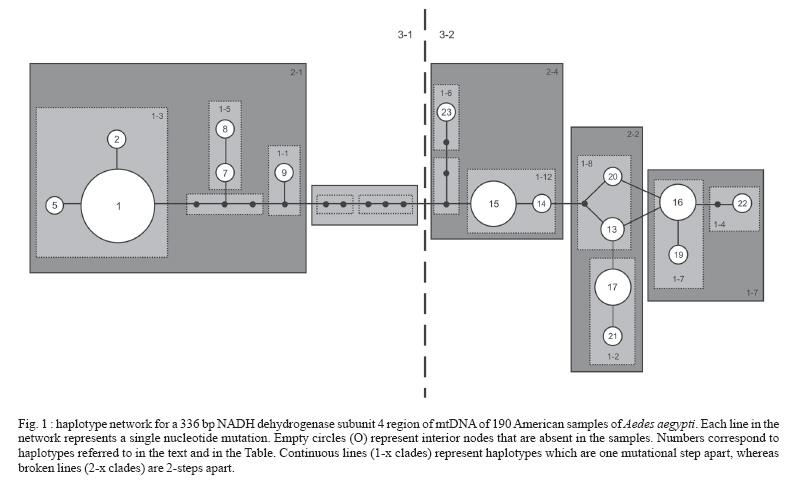
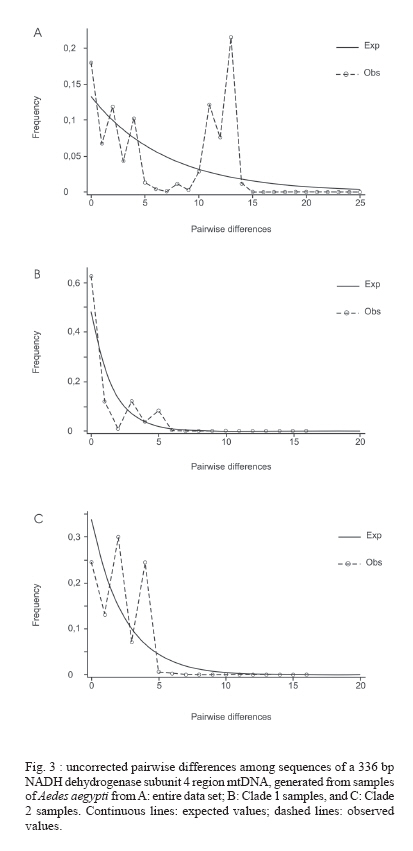
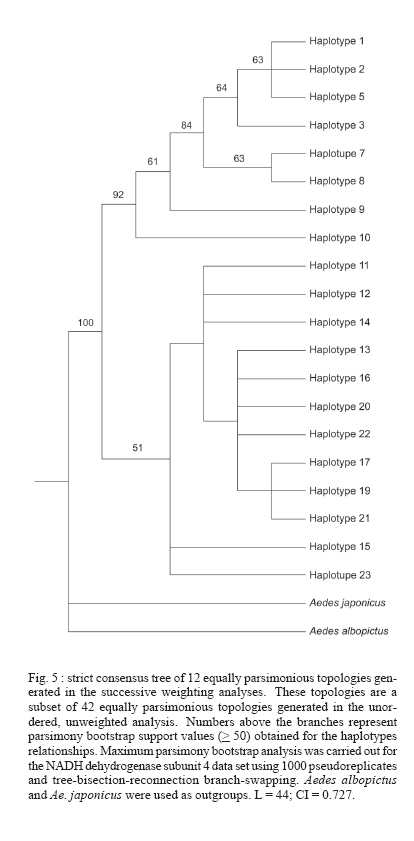
Paulo, Av. Dr. Arnaldo 715, 01246-904 São Paulo, SP, Brasil Financial support: Sucen, Fapesp (00/12138-7; 06/01816-0), CNPq (501529/2003-9) Received 30 November 2006 Code Number: oc07091 To analyze the genetic relatedness and phylogeographic structure of Aedes aegypti, we collected samples from 36 localities throughout the Americas (Brazil, Peru, Venezuela, Guatemala, US), three from Africa (Guinea, Senegal, Uganda), and three from Asia (Singapore, Cambodia, Tahiti). Amplification and sequencing of a fragment of the mitochondrial NADH dehydrogenase subunit 4 gene identified 20 distinct haplotypes, of which 14 are exclusive to the Americas, four to African/Asian countries, one is common to the Americas and Africa, and one to the Americas and Asia. Nested clade analysis (NCA), pairwise distribution, statistical parsimony, and maximum parsimony analyses were used to infer evolutionary and historic processes, and to estimate phylogenetic relationships among haplotypes. Two clusters were found in all the analyses. Haplotypes clustered in the two clades were separated by eight mutational steps. Phylogeographic structure detected by the NCA was consistent with distant colonization within one clade and fragmentation followed by range expansion via long distance dispersal in the other. Three percent of nucleotide divergence between these two clades is suggestive of a gene pool division that may support the hypothesis of occurrence of two subspecies of Ae. aegypti in the Americas. Key words: phylogeography - Aedes aegypti - NADH dehydrogenase subunit 4 - nucleotide divergence The pandemic of dengue fever (DF) and dengue hemorrhagic fever (DHF) is one of the most important public health problems in the modern world, especially in tropical countries (WHO 2004). Because there is no vaccine against dengue viruses (DENV), most efforts to manage the disease are based on vector control. Consequently, information about genetic variability, population structure, and migration dynamics of the major urban vector of DENVs, Aedes aegypti, could prove valuable for the design of effective control strategies. Ae. aegypti is an autochthonous African species which arrived in the Americas aboard slave ships and became established and spread throughout the New World in the XVII and XVIII centuries (OPAS 1995). By the 1950s, all countries in the Americas except Canada registered the presence of Ae. aegypti. Abundance and distributional range of the vector were reduced from the 1940s through the 1970s as a result of the Ae. aegypti eradication program adopted by the Pan American Health Organization (PAHO) (Franco 1961). However, the PAHO vector control program was discontinued in the 1960s and, consequently, Ae. aegypti re-infested all countries from which it was supposed to have been eliminated (Schatzmayr 2000), once again becoming a health concern. The genetic variability of Ae. aegypti in the Americas has probably been greatly reduced because of a bottleneck effect during its introduction and selective pressure resulting from a strong vector control policy that focused mainly on Ae. aegypti control similarly to what occurred to Anopheles mosquitoes in Central and South America (Donnelly et al. 2002). The presence of Ae. aegypti in Asia was first reported in the XIX century (Smith 1956); however, it is possible that it had been introduced earlier, either in the XVII or XVIII century, due to the trade between African and Asian countries. Currently the species is spread throughout the world, in tropical and subtropical areas from 45º N to 35º S (Slosek 1986). The main objectives of the present study were: (1) to examine the distribution of the mitochondrial lineages of populations of Ae. aegypti collected in American, African, and Asian countries and (2) to estimate the phylogenetic relationships among the Ae. aegypti haplotypes. Three hundred thirty-six nucleotide base-pairs of the mitochondrial NADH dehydrogenase subunit 4 (ND4) region were used. MATERIALS AND METHODS Details about collection locations, dates, and sample size of Ae. aegypti populations used in this study are in the Table and available upon request from the corresponding author. Mosquito eggs were collected in ovitraps placed in several urban locations in Brazil, Venezuela, and the US. Eggs were raised to adult stages and a random sample of the adults was identified to species and preserved in 100% ethyl alcohol maintained at -70º C. Adult mosquitoes from Cambodia, Guatemala, Guinea, Peru, Senegal, Singapore, Uganda, and Tahiti were obtained from collaborating researchers. Genomic DNA of individual adults was obtained by phenol-chloroform extraction (Sambrook et al. 1989) and eluted in 100 ml of TE buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0). Primers used to amplify and sequence a 336 bp portion of the mitochondrial ND4 gene were: ND4F: 5-ATT GCC TAA GGC TCA TGT AG-3 and ND4R: 5-TCG GCT TCC TAG TCG TTC AT-3. Polymerase chain reactions (PCR) were carried out in a total volume of 50 µl using standard protocols, with 2 mM Mg++, 20 mM Tris-HCl (pH 8.4), 50 mM KCl, 0.5 mM of each primer, 0.2 mM dNTP mix, 2 U Taq DNA polymerase (Invitrogen), and 1 µl of template DNA. Each experiment a negative control was performed. PCR temperature profiles to obtain the above sequence were: initial denaturation at 94ºC for 2 min followed by 35 cycles at 94ºC for 1 min, 56ºC for 30 s, and 72ºC for 1 min, and final extension at 72ºC for 7 min. For sequencing, PCR products were purified using PEG precipitation [20% polyethylene glycol (PEG) 8000/2.5 M NaCl]. Sequencing reactions were carried out directly on both strands of DNA using ABI Big Dye chemistry (PE Applied Biosystems, Foster City, CA) and the sequences generated with an ABI 377 automated sequencer. The amplified ND4 region corresponds to 336 base pairs from position 8488 to 8823 in the Anopheles gambiae mitochondrial genome (GenBank accession number L20934). Sequences were edited using BioEdit Sequence Alignment Editor program (Hall 1999), and aligned in CLUSTAL X (Thompson et al. 1997). Sequence similarity of the ND4 sequences generated in this study with those previously available in GenBank was assessed using BLASTA search (Altschul et al. 1990). Tajima's D test (Tajima 1989) and F* and D* tests (Fu & Li 1993) of selective neutrality were carried out using the DnaSP software (Rozas et al. 2003). The same software was employed to estimate the significance levels of these estimates using a distribution of 10,000 simulated populations generated by the neutral coalescent process. DnaSP was also used to (1) estimate the genetic diversity parameters haplotype diversity (Hd), nucleotide diversity (p), and the average number of nucleotide differences (k) for all data sets and (2) the average number of nucleotide differences (k) between clades. To test the role of ecological and/or historical events on the genetic structure of American populations, a minimum spanning network of Ae. aegypti haplotypes was created using TCS 1.18 (Clement et al. 2000). GeoDis 2.0 (Posada et al. 2000) was used to determine the statistical significance, after 10,000 replicates, of the geographic association between haplotypes. Templeton's inference key (Templeton 2004) was used to determine the likely mechanism of the observed genetic structure. Mismatch distribution was estimated employing Arlequin 2000 (Schneider et al. 2000) using a generalized non-linear least-square approach (Schneider & Excoffier 1999). Phylogenetic relationships among all Ae. aegypti populations were inferred with Network 4.2.0.1 (Fluxus Technology Ltd. at www.fluxus-engineering.com) using the median joining method (Bandelt et al. 1999). Unweighted parsimony was performed in PAUP 4.0b10 (Swofford 2002) using a heuristic search with tree-bisection-reconnection (TBR) and 1000 random-taxon additions. Parsimony bootstrap support values were generated from 1000 pseudoreplicates with 10 random-taxon-addition replicates per pseudoreplicate. Parsimony uninformative characters were excluded from all the analyses. Sequences of Ae. albopictus (GenBank accession number NC006817) and Ae. japonicus (GenBank accession number AF305879) were employed as outgroups. RESULTS Three hundred thirty-six base-pairs long sequences from 218 individuals were obtained and used for phylogenetic and population genetic analyses (190 American samples and 28 from other countries). Twenty haplotypes were obtained (GenBank accession numbers DQ176828 to DQ176831, DQ176833 to DQ176843, and DQ176845 to DQ176849): 14 haplotypes were exclusive to the Americas, four exclusive to African/Asian countries, one is common to the Americas and Africa, and one to the Americas and Asia (Table). The haplotypes showed 22 nucleotide substitutions in 336 base-pairs, 18 synonymous substitutions and four non-synonymous substitutions. Tajima's D test yielded a value of 2.09516 (not significant, P > 0.05), whereas Fu & Li's D* and F* values were -0.62927 (not significant, P > 0.1) and 0.55971 (not significant, P > 0.1), respectively. Based on these results, we assumed an infinite sites model (Bertorelle & Slatkin 1995) in subsequent analyses. Statistical parsimony (Templeton et al. 1992, Posada et al. 2000) was used to generate a network for 16 haplotypes sampled in the American populations of Ae. aegypti. Relationships among distinct haplotypes are presented in Fig. 1, which shows two major haplogroups, designated clades 1 and 2. These two clades are separate by eight mutational steps. Clade 1 is composed of a frequent haplotype designated haplotype 1, which is connected to other less frequent haplotypes (2, 5, 7, 8, 9) by one or a few mutational steps. Clade 2 includes haplotypes 13, 14, 15, 16, 17, 19, 20, 21, 22, 23. This clade shows a complex evolutionary pattern, with reticulation and several high-frequency haplotypes. For the entire data set the estimated haplotype diversity (Hd) was 0.82, the nucleotide diversity (p) under the Jukes and Cantor model was 0.01997, and the mean number of nucleotide differences (k) was 6.54779. The haplotype diversity for Clade 1 was 0.3750 and for Clade 2 it was 0.75483, whereas the nucleotide diversity under the Jukes and Cantor model was 0.00325 and 0.00590 for clades 1 and 2, respectively. Significant Dc and Dn distance values are shown in Fig. 2. The nested clades 1-12, 2-1, 3-2, and the total cladogram revealed significant Dc and Dn values, although the tip/interior status could not be determined for Clade 1-12 and the total cladogram and thus inferences here were inconclusive. Results suggested long distance colonization via long distance dispersal for group 2-1 (which is Clade 1) and fragmentation followed by range expansion for nested clade 3-2 (Clade 2) (Figs 1, 2). Pairwise mismatch distributions revealed a multi-modal distribution for the entire data set and Clade 2 (Fig. 3A,C), whereas a unimodal distribution, very similar to expected values to a mutation-drift equilibrium model, was observed in Clade 1 haplotypes (Fig. 3B). Phylogenetic relationships were estimated using both median-joining networks (MJN) and maximum parsimony (MP). As a result MJN and MP topologies show two major groups, which are separated by six mutational steps: Senegal haplotypes (3, 10) are related to Clade 1 whereas Asian and Guinea haplotypes (11, 12, 13, 23) are related to Clade 2 (Figs 4, 5). American haplotypes were recovered in both Clade 1 and Clade 2. DNA divergence between clades 1 and 2 showed an average number of nucleotide differences of 12.015, a persites divergence with Jukes and Cantor (K) between clades of 0.03207. DISCUSSION Results of the genetic analyses showed two Ae. aegypti mitochondrial lineages in the Americas: one related to African (Senegal) haplotypes and the other to African (Guinea and Uganda) and Asian haplotypes. Similarly, Powell et al. (1980) studied the genetic variability of allozyme loci from 34 Ae. aegypti aegypti populations and showed that Caribbean, South American, and US populations were genetically related to those of Ae. aegypti aegypti from Africa, while populations of Ae. aegypti aegypti from Asia were close to Ae. aegypti formosus. They also proposed that the African-related Ae. aegypti aegypti population had been introduced into the Americas and Caribbean during the XVII and XVIII centuries. The nucleotide diversity found in the present study and in Mexican populations (Gorrochotegui-Escalante et al. 2000) were three to eight times higher than those registered for An. gambiae (p = 0.0038) and An. arabiensis (p = 0.0023 0.0051) (Besansky et al. 1997). However, when estimated separately, the p value for Clade 1 (0.00325) and for Clade 2 (0.00590) were similar to those found for the Anopheles sibling species, suggesting that Ae. aegypti clades 1 and 2 evolved as two independent genetic lineages. Likewise, the nucleotide divergence (K) between clades 1 and 2 (0.03207) is similar to that found for putative species. For instance, the divergence estimated in a RFLP analysis of mitochondrial DNA in populations of An. nuneztovari from the Brazilian Amazon and Western Colombia (0.033 to 0.037) suggested that these Anopheles geographic populations constitute separate species (Scarpassa et al. 2000). Within the Drosophila group, the estimated mtDNA divergences were 0.034 and 0.039 between D. melanogaster and the sibling species D. simulans and D. mauritiana, respectively. Comparisons between D. simulans and D. mauritiana showed that the estimated mtDNA divergence was 0.012 (Caccone et al. 1988). The NCA analysis revealed that the origin of this structure is historical. The nested clade 2-1 (Clade 1) showed a long distance colonization pattern. Haplotypes 1, 2, and 5 which clustered in nested clade 1-3 (Fig. 1), may be a recent diversification of haplotype 1, which could be a South American haplotype remnant of the eradication program. The star shape of the Brazilian content of Clade 1 (mainly nested clade 1-3, Fig. 1) and the unimodal, relatively smooth and narrow-tailed mismatch distribution of pairwise differences, very similar to expected values (Fig. 3B), supports a hypothesis that Clade 1 experienced a recent demographic expansion (de Brito et al. 2002, Slatkin & Hudson 1991). Groups 1-1 and 1-5 could be an introduction of US and/or Venezuelan haplotypes with a secondary introduction probably through Brazilian harbors, leading to the long distance movement pattern showed by the nested clade 2-1. The pattern displayed by nested clade 3-2 (Clade 2, Fig. 2) show a fragmentation that was followed by range expansion by long distance dispersal. This pattern maybe explained by a fragmentation of the Asian populations that was followed by a colonization of the Americas. Nested clade 3-2 is more polymorphic and shows a complex structure. The presence of reticulation among the haplotypes may be a consequence of multiple introductions of Ae. aegypti or ancient polymorphism. Phylogenetic analyses of the ND4 haplotypes employing the MJN (Fig. 4) and MP (Fig. 5) criteria recovered of the same two clades. MP bootstrap support for clades 1 and 2 is moderate (92%). Results of SP and MP analyses showed that the haplotypes from West Africa (Senegal) and those from the Americas (Brazil, Peru, Venezuela, and US) are closely related. Interestingly, Ae. aegypti has never been eliminated from the US or Venezuela. Perhaps, individuals from one or more of these countries could have infested Brazil after the eradication of the 1950s (if the eradication really happened). The haplotypes included in Clade 2 are closely related to those found in West Africa (Guinea), East Africa (Uganda), Asia (Singapore, Cambodia, Tahiti), and the Americas. Similar results were found in a study using Mexican populations of Ae. aegypti and the same ND4 gene region. Gorrochotegui-Escalante et al. (2000) found two genetic clusters. The presence of two genetically related groups in Mexico was corroborated by the results of maximum likelihood analyses (ML) that employed a broad sample size (Gorrochotegui-Escalante et al. 2002). Furthermore, results of the MP and ML analyses using a combined data set that included the data employed in the current study plus several Mexican sequences downloaded from the GenBank generated the same two clusters (not shown). Recent studies on populations of Ae. aegypti from Venezuela and Peru, using the same fragment of the ND4 gene and MP analyses, generated two clusters (da Costa-da-Silva et al. 2005, Herrera et al. 2006). Moreover, in both studies, the results of the AMOVA demonstrated the major component of variation to be within population. This result suggests that the two clades occur in sympathy. A similar result was found in the current study. Results of all the analyses support the hypothesis that the mitochondrial lineages of Ae. aegypti aegypti were introduced into the Americas during the XVII and XVIII centuries, and experienced a bottleneck caused by the Ae. aegypti vector control measures adopted in the 1950s and 1960s. During the 1970s, Ae. aegypti re-infested most American countries. Two evolutionary scenarios may explain this. First, it is possible that some individuals survived the intensive vector control program adopted by the American countries. A second scenario is that of dispersion of Ae. aegypti from populations that remained in those countries where eradication of Ae. aegypti had never been achieved. Consequently, an old lineage of Ae. aegypti persevered and dispersed throughout the Americas. Clade 1 haplotypes may be representative of those lineages. Additionally, it is possible that Clade 2 haplotypes were introduced into the Americas during the 1980s as a consequence of the intense commercial exchanges with Asian countries and the commercial globalization. Similarly, the dispersion of Ae. albopictus from Asian countries to the Americas occurred during the same period of time and was caused by the intense commercial exchange (Gubler 2003). We raised the hypothesis that the haplotypes included in Clade 2 were introduced into the Americas from Asia countries with which the commercial exchanges are intense. As a result, the haplotypes that formed Clade 2 were transported to other regions along commercial routes. We speculate that the two mtDNA strains found by Powell et al. (1980) met each other in the Americas, after the 1980s. The recent finding of two separated clusters in Asian populations (Mousson et al. 2005, Bosio et al. 2005) could be a result of a secondary introduction of lineages from the Americas into Asia, or that an evolutionary force is maintaining the observed mtDNA lineages, similar to an incipient speciation process. The introduction of new lineages has an enormous epidemiological importance because different populations of Ae. aegypti may present distinct susceptibility to both DENV and yellow fever virus (YFV) infections and to insecticides. Failloux et al. (2002) showed that genetic differences among Ae. aegypti populations depend on colonization history and may be associated with susceptibility to DENV-2. Lourenço-de-Oliveira et al. (2002) demonstrated the Brazilian populations of Ae. aegypti to be heterogeneous to susceptibility to both the YFV and DENV-2 infection. Additionally, lineages of Ae. aegypti may respond different to vector control measures when these make use of insecticides. The pattern of the mtDNA haplotype diversity is consistent with two genetically distinct lineages of Ae. aegypti, and are suggestive that there might be two sub-species circulating in the Americas. Further studies will be necessary to test this hypothesis. ACKNOWLEDGMENTS To Sergio Russo Matiolli and Flora Fernandes-Matiolli for their discussion of the points we have raised; to Kleber José Whitaker and Alexandre Moreira for their laboratory support. To the three anonymous reviewers for their contributions to improve a first version of the manuscript. REFERENCES
Copyright 2007 Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc07091f2.jpg] [oc07091t1.jpg] [oc07091f1.jpg] [oc07091f3.jpg] [oc07091f5.jpg] [oc07091f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}