 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
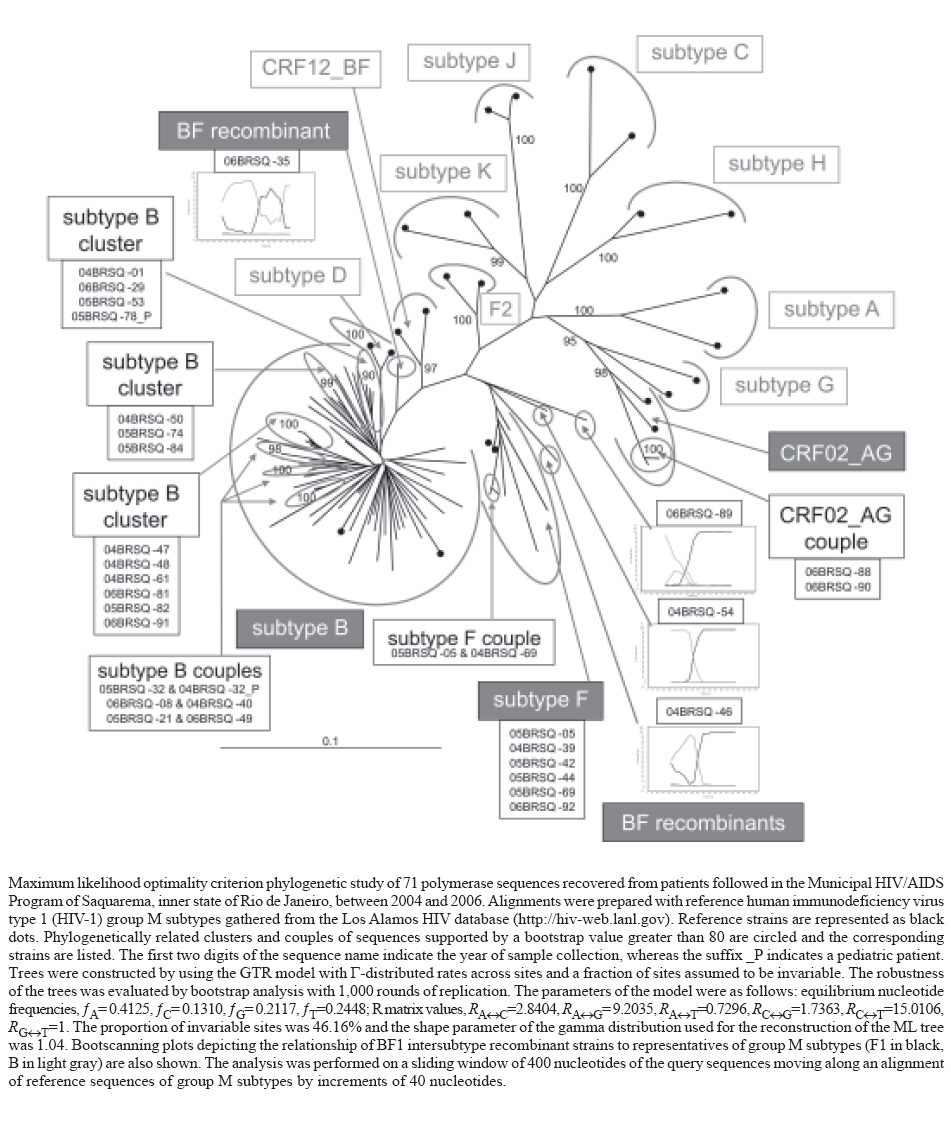
Memórias do Instituto Oswaldo Cruz, Vol. 102, No. 7, November, 2007, pp. 809-815 Autochthonous horizontal transmission of a CRF02_AG strain revealed by a human immunodeficiency virus type 1 diversity survey in a small city in inner state of Rio de Janeiro, Southeast Brazil Walter A Eyer-Silva/*/**/+, Mariza G Morgado Laboratório
de Aids & Imunologia Molecular, Instituto Oswaldo Cruz-Fiocruz,
Av. Brasil 4365, 21045-900 Rio de Janeiro, RJ, Brasil *Hospital
Universitário Gaffrée e Guinle, Universidade Federal
do Estado do Rio de Janeiro, Rio de Janeiro, RJ, Brasil **Programa
Municipal de HIV-1/Aids de Saquarema, Saquarema, RJ, Brasil Received
27 February 2007 Code Number: oc07147 As part of an ongoing study on the features of AIDS spread towards small cities and rural areas, we present a molecular survey of human immunodeficiency virus type 1 (HIV-1) polymerase sequences recovered between 2004 and 2006 from 71 patients receiving care in the city of Saquarema, inner state of Rio de Janeiro. Phylogenetic reconstructions found the two prevalent lineages in the state (subtypes B [59 strains, 83.1%], F1 [6 strains; 8.4%], and BF1 recombinants [four strains; 5.6%]), as well as two (2.8%) CRF02_AG strains, which seems to be an emerging lineage in the capital. These CRF02_AG sequences were recovered from a married heterosexual couple who never traveled abroad, thus providing the first molecular evidence of autochthonous horizontal transmission of this lineage of major global importance. Also, three phylogenetic clusters of strains recovered from a total of 18.3% of the cohort were uncovered. Their close genetic relatedness suggests they were recovered from patients who probably took part in the same chain of viral spread. In conjunction with our previous surveys from inner Rio de Janeiro, these results suggest that although small cities harbor unique molecular features of HIV-1 infection, they also clearly reflect and may rapidly absorb the diversity recorded in large urban centers. Key words: Brazil - CRF02_AG - human immunodeficiency virus type 1 - molecular epidemiology - transmission clusters The extremely high genetic diversity of human immunodeficiency virus type 1 (HIV-1) has been successfully used to study the regional and global distribution of different viral groups and subtypes (Hu et al. 1996) and makes it possible to trace patterns of viral spread between populations, groups and people (Kuiken et al. 2000). Phylogenetic analysis allows classification of HIV-1 strains into three groups: M, N and O. The M (main) group, the widest spread, has been further subdivided into at least nine subtypes (A-D, F-H, J, and K) and 32 circulating recombinant forms (CRF), whereas a high diversity of unique intersubtype recombinant forms has also been demonstrated (Leitner et al. 2005). In Brazil, molecular analyses of HIV-1 nucleotide sequences have shown an epidemic driven mainly by three group M subtypes: B, C, and subsubtype F1 (heretofore designated subtype F1), as well as a myriad of unique BF1 intersubtype recombinants (Morgado et al. 1998, Tanuri et al. 1999, Vicente et al. 2000, Brindeiro et al. 2003, Soares et al. 2003). Isolated cases of other subtypes, such as subtype D (Morgado et al. 1998, Couto-Fernandez et al. 2006), subtype A (Caride et al. 2000), CRF02_AG (Pires et al. 2004, Couto-Fernandez et al. 2005), CRF28_BF and CRF29_BF (De Sa Filho et al. 2006) have also been reported. Phylogenetic analyses of HIV-1 nucleotide sequences are being increasingly used to better understand epidemiological patterns of viral spread. Such studies have proven to be invaluable to clarify the routes of viral transmission in community settings (Holmes et al. 1995, Brown et al. 1997), to establish (Ou et al. 1992, Blanchard et al. 1998) or rule out (Holmes et al. 1993, Jaffe et al. 1994) iatrogenic viral transmission, to study the pathways of viral spread among clusters of patients (Leitner et al. 1996, Robbins et al. 2002), to trace the geographic origin of recently introduced subtypes (Gao et al. 1996, Kato et al. 1999, Yu et al. 2001, Couto-Fernandez et al. 2006), to infer the demographic history and date of origin of epidemic spread (Robbins et al. 2003, Bello et al. 2006), to investigate unusual forms of viral transmission (Goujon et al. 2000, Andreo et al. 2004), and has also found forensic applications (Albert et al. 1994, Banaschak et al. 2000, Metzker et al. 2002, Lemey et al. 2005). Phylogenetic analyses of HIV-1 sequences have also been used to provide evidence of a high incidence molecular profile, as evidenced by multiple transmission clusters, low level of sequence diversity and signature amino acid substitutions (Liitsola et al. 1998, Oelrichs et al. 2000, Shankarappa et al. 2001, Nguyen et al. 2002), and of an old and mature epidemic pattern, given the high sequence diversity and the absence of transmission clusters (Vidal et al. 2000, Trask et al. 2002). It is known that the AIDS epidemic in Brazil is spreading from the large urban centers towards small cities and the innermost parts of the country (Szwarcwald et al. 2000). Therefore, in an effort to better understand HIV-1 diversity, the molecular epidemiological patterns, and to assess the existence of micro-epidemic lineages associated with HIV-1 spread towards inner Brazil, our group is carrying out cross-sectional surveys in selected, small inner cities of the state of Rio de Janeiro. We have recently reported on the molecular epidemiology of HIV-1 infection in two small, neighboring cities in the northwestern region of the state: Miracema and Santo Antonio de Pádua (Eyer-Silva & Morgado 2006, Eyer-Silva et al. 2007a, b). We found both a polyphyletic pattern suggestive of multiple viral introductions in the region, and that more than 40% of the recovered sequences fell within clusters of genetically related strains, suggesting the existence of sexual networks and a high incidence molecular profile. Additionally, phylogenetic clusters from one city were unrelated to those found in the other, suggesting that each of these two neighboring cities harbors its own set of micro-epidemic lineages (Eyer-Silva et al. 2007a). We now expand our molecular characterization of HIV-1 strains to another city in inner state of Rio de Janeiro: Saquarema, in the Sun Coat Region. The data presented herein shall broaden our understanding of the genetic diversity and the molecular epidemiological profile associated with HIV-1 spread in inner state of Rio de Janeiro. PATIENTS, MATERIALS AND METHODS Patients and setting - Saquarema is situated 116 km north from the city of Rio de Janeiro and counted, as of July 2005, 61,591 inhabitants distributed in a geographic area of 355 km2 (http://www.ibge.gov.br/cidadesat/). Bathed by the Atlantic, the city enjoys a nationwide reputation for its natural beauty and attracts thousands of domestic and foreign visitors and vacationers every year. A municipal HIV/ AIDS program was established in 2000 and patients are offered regular medical appointments. The present study protocol was reviewed and approved by the Ethics Review Board at Instituto de Pesquisa Clínica Evandro Chagas, Fundação Oswaldo Cruz, Rio de Janeiro. Nucleotide sequencing and phylogenetic reconstructions - After signed informed consent, blood samples were collected, DNA was extracted from peripheral blood mononuclear cells, and the sequence of a single fragment (around 1000 nucleotides) of HIV-1 polymerase gene encompassing the whole protease (PR) and reverse transcriptase (RT) amino acids 1-230 was generated using a previously described in-house protocol (Eyer-Silva & Morgado 2005). In spite of being a relatively conserved region of the HIV-1 genome, the polymerase gene has been shown to contain enough phylogenetic signal for subtyping analysis (Cornelissen et al. 1997) and to allow studies of epidemiologic relationships between populations, groups and people (Hue et al. 2004, Eyer-Silva & Morgado 2006). Sequences were deposited on the GenBank database under accession numbers DQ899721 through DQ899721 and EF042627 through EF042690. Sequences were aligned against a set of reference strains from HIV-1 group M subtypes and trimmed to equivalent lengths by using CLUSTAL X (Thompson et al. 1997). Minor adjustments were performed using BioEdit (Hall 1999). An alignment of 901 nucleotides, corresponding to positions 2313 through 3213 relative to HXB2 genome, was obtained. Phylogenetic inferences were performed by applying the maximum likelihood (ML) optimality criterion (Felsenstein 1973) as implemented in PAUP* version 4.0b10 (Swofford 1999). The best fitting nucleotide substitution model was selected by using the Akaike criterion as implemented in MODELTEST version 3.06 (Posada & Crandall 1998). We found the GTR model with gamma-distributed rates across sites and a fraction of sites assumed to be invariable to be the best fitting. The parameters of the chosen model are indicated in the Figure. ML trees were run with PAUP* using heuristic searches based on a subtree pruning and regrafting (SPR) branch-swapping algorithm (Felsenstein 2004) and 1000 rounds of bootstrap replication (Felsenstein 1985). The robustness of the tree was evaluated by bootstrap analysis with 1,000 rounds of replication and the phylogenetic tree was schematically represented with the TreeView program (Page 1996). The bootscanning method was used to detect and study recombination, as implemented in the SIMPLOT software, version 3.5 (Salminen et al. 1995). We found no evidence of hypermutation in the dataset when sequence CD83ELI (GenBank accession no. K02454) was used as the reference strain in the HYPERMUT program (Rose & Korber 2000). The presence of nucleotide substitution saturation at different codon positions was analyzed by plotting the transitions and transversions versus the F84 model of substitution by using the DAMBE software (Xia & Xie 2001). No nucleotide substitution saturation could be observed on inspection of the plots. This implies that the dataset was phylogenetically informative. Statistical analyses - Patients whose samples yielded sequences that fell within a phylogenetic cluster were compared with those whose samples yielded non-clustered sequences. Depending on the variables, the Wilcoxon rank sum test, and the Fisher's exact test were used for the univariable analysis. Multivariate logistic regression was used to identify variables independently associated with being a patient whose sample yielded clustered sequences (response variable). Age, gender, place of birth and residence, marital status, exposure category, AIDS-defining CDC stage, having a known direct epidemiologic relationship within the cohort, having always lived in Saquarema, previous use of snorted cocaine, and level of education were studied as explanatory variables. Statistical analyses were performed using the software R (Ihaka & Gentleman 1996). RESULTS Assignation to subtypes - Between October 2001 and May 2006 a total of 93 HIV-infected adult patients received care at a local ambulatory facility. A detailed clinical and epidemiological characterization of this cohort will be presented elsewhere. Between the years of 2004 and 2006 blood samples were collected from 73 enrolled patients. A total of 71 strains were recovered from 68 adult and three pediatric patients (including one mother and daughter couple). We failed to obtain sequences from two samples. The Figure presents the ML phylogenetic reconstruction. A total of 59 sequences (83.1%) were assigned group M subtype B. The remaining sequences were assigned group M subtypes F1 (6 strains; 8.4%), CRF02_AG (2 strains; 2.8%), and unique BF1 recombinants (4 strains; 5.6%). Three unique BF1 recombinants (04BRSQ-46, 04BRSQ-54, 06BRSQ-89) harbored a subtype B fragment in the PR region and a subtype F1 fragment in the RT gene. The remaining BF1 mosaic strain (06BRSQ-35) displayed a predominant subtype B pattern with an F1 fragment in the RT region (Figure). Phylogenetic analyses of these BF1 recombinants found that they are not related to previously described CRF_BF (data not shown). Identification of phylogenetically related clusters and couples - ML phylogenetic reconstruction yielded three subtype B clusters encompassing a total of 13 strains (Figure), corresponding to 18.3% of all generated sequences. The largest cluster harbored six sequences and was supported by a bootstrap value of 100. Two other subtype B clusters harbored four and three strains and were supported by bootstrap figures of 90 and 99, respectively. The former included a pediatric patient (05BRSQ-78_P) whose mother had died of complications of AIDS before the study period. Additionally, five phylogenetically-related couples of sequences were also seen: three couples were assigned subtype B, whereas the other two were assigned subtype F and CRF02_AG. These CRF02_AG strains (06BRSQ-88 and 06BRSQ-90) branched with a bootstrap value of 100 and were recovered from a married heterosexual couple who reported never have traveled abroad. Patient 06BRSQ-88, a 28-year-old male, attributed the acquisition of HIV-1 infection to unprotected sexual contacts in the city of Rio de Janeiro. His 29-year-old wife (patient 06BRSQ-90) reported having always lived in Saquarema. Known epidemiologic relationships in the cohort - All sequences recovered from patients whose epidemiologic relationships were known a priori were found to be phylogenetically associated within one of the observed clusters or couples of related strains. These were the five phylogenetically-related couples (patients SQ-32/SQ-32_P, the mother and child couple, SQ-88/SQ90, a married couple infected with CRF02_AG strains, as well as couples SQ-01/SQ-53 and SQ-81/SQ-82 and the triad SQ-47/SQ-48/SQ-61), all of whom found to be infected with clustered sequences. The other patients whose sequences were found to be phylogenetically associated had no known direct epidemiologic relationship. Demographic, social, clinical and behavioral associ-ations within phylogenetic clusters - Samples from 12 (17.6%) out of 68 adult patients yielded clustered sequences. On univariable analysis, these patients were found to be younger, more likely to have a known epidemiologic link within the cohort, to always have lived in Saquarema and to reside within the geographic limits of Saquarema as of sampling. Multivariable logistic regression analysis identified having a known direct epidemiologic relationship (odds ratio [OR] 9.44, 95% confidence interval [CI] 7.83-11.05, P = 0.006) and having always lived in Saquarema (OR 9.11, 95% CI 7.29-10.93, p = 0.017) as independent predictors of belonging to a cluster. The present molecular survey of HIV-1 polymerase sequences from the city of Saquarema uncovered the occurrence of three HIV-1 subtypes (B, F1, and CRF02_AG) and four BF1 mosaic forms. It is important to highlight that these subtype assignments have been made based on the study of a fragment of the polymerase gene and can not be extrapolated to other viral genomic regions. The finding of such a genetic diversity in a small city in inner state of Rio de Janeiro further highlights the complexity of the molecular epidemiology of HIV-1 in Brazil. Additionally, we present the first molecular evidence of horizontal transmission of subtype CRF02_AG in Brazil. This is a rare HIV-1 variant in the country and only two cases have been published in peer-reviewed journals (both from the metropolitan area of Rio de Janeiro) (Pires et al. 2004, Couto-Fernandez et al. 2005). However, the recent identification of novel cases of HIV-1 infection due to CRF02_AG at our laboratory (unpublished observations) suggests that this might prove to be an emerging lineage in the city of Rio de Janeiro. The present molecular evidence of autochthonous heterosexual transmission of CRF02_AG strains, an HIV-1 variant of major global importance, emphasize the importance of continued monitoring of viral diversity in Brazil. Phylogenetic analyses found evidence of three clusters of subtype B strains recovered from a total of 12 adult patients and one pediatric case whose mother had died of complications of AIDS before the study period. The largest cluster harbored six strains, whereas two other clusters contained four and three sequences (Figure). These intracluster strains shared close genetic relatedness and were phylogenetically associated with high bootstrap values, suggesting that they were recovered from patients who probably took part in the same chain of viral spread. Similar phylogenetic clusters have also been identified in our previous molecular epidemiology surveys of HIV-1 strains from two small cities in northwestern state of Rio de Janeiro. In the present study, however, we found a lower prevalence of clustered sequences. Whereas in the previously reported analyses more than 40% of the strains were found to belong to a phylogenetic cluster (Eyer-Silva & Morgado 2006, Eyer-Silva et al. 2007a), in the present Saquarema study 13 (18.3%) of the strains fell within a cluster. The lower prevalence of phylogenetically related cases probably is due to differences in the characteristics of the cohorts. When compared to the two northwestern cities, patients from Saquarema were significantly more likely to have been born elsewhere and to have initiated HIV follow up in another city (data not shown). For instance, 60.2% of patients from Saquarema were born elsewhere, whereas the corresponding figures for Santo Antonio de Pádua and Miracema were 20.6% and 11.1%, respectively (p < 0.01). In agreement with our previous studies (Eyer-Silva & Morgado 2006), patients whose sequences fell within a cluster were significantly more likely to have a known epidemiologic relationship within the cohort and to always have lived in the studied city. In Brazil relatively little attention is being paid to the study of the features of the spread of the AIDS epidemic towards small cities and rural areas. This molecular analysis of HIV-1 sequences from the small city of Saquarema, in conjunction with our previous surveys from the inner cities of Miracema and Santo Antonio de Pádua, demonstrate the cocirculation in inner state of Rio de Janeiro of the two prevalent subtypes in Southeast Brazil (B, F1, and BF1 recombinants), as well as two minor variants previously recorded in the capital: CRF02_AG, presented in this report, and the recently studied subtype D (Morgado et al. 1998, Couto-Fernandez et al. 2006, Eyer-Silva et al. 2007a). Moreover, we present the first molecular evidence of autochthonous horizontal transmission of CRF02_AG strains in Brazil. These results suggest that although small cities of inner state of Rio de Janeiro harbor unique molecular features of HIV-1 infection, they also clearly reflect and may rapidly absorb the diversity recorded in large urban centers. The finding of such a complex pattern of viral subtypes and recombinant forms in small cities of Rio de Janeiro also underscore the need for a continued study of the diversity and routes of HIV-1 spread in inner Brazil. These surveys will lead to a better understanding of the dynamics of the Brazilian HIV-1 epidemic, will be invaluable to monitor the prevalence and emergence of different viral lineages and may eventually be important to geographically related vaccine development. ACKNOWLEDGEMENTS To the patients followed at the Municipal HIV/ AIDS Program of Saquarema, for agreeing to participate in this study. To the many dedicated health care professionals in Saquarema, without whom this study would not be possible. REFERENCES
Copyright 2007 Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc07147f1.jpg] |
| |||||||||

{kind=link}