 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 103, No. 2, March, 2008 , pp. 119-129 The hope for an HIV vaccine based on induction of CD8+ T lymphocytes - A Review David I Watkins Department
of Pathology and Laboratory Medicine, University of Wisconsin-Madison,
555 Science Dr., 53711 Madison, WI, USA Financial support: National Institutes of Health (grants R01 AI049120, R01 AI052056, R24 RR015371 and R24 RR016038) Received
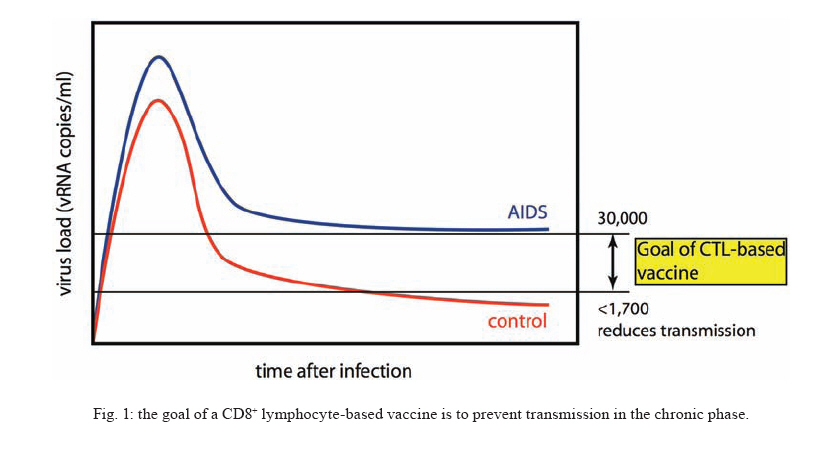
12 March 2008 Code Number: oc08022 The only long-term and cost-effective solution to the human immunodeficiency virus (HIV) epidemic in the developing world is a vaccine that prevents individuals from becoming infected or, once infected, from passing the virus on to others. There is currently little hope for an AIDS vaccine. Conventional attempts to induce protective antibody and CD8+ lymphocyte responses against HIV and simian immunodeficiency virus (SIV) have failed. The enormous diversity of the virus has only recently been appreciated by vaccinologists, and our assays to determine CD8+ lymphocyte antiviral efficacy are inadequate. The central hypothesis of a CTL-based vaccine is that particularly effective CD8+ lymphocytes directed against at least five epitopes that are derived from regions under functional and structural constraints will control replication of pathogenic SIV. This would be somewhat analogous to control of virus replication by triple drug therapy or neutralizing antibodies. Key words: CTL - HIV - SIV - Vaccines The problems with contemporary human immunodeficiency virus (HIV) vaccines The failure of the AIDSVAX HIV vaccine was announced in 2003, showing that Env-specific vaccine-induced antibodies that do not neutralize will not prevent infection. A canarypox-based vaccine trial started in the fall of 2003, amidst considerable controversy as to its likely efficacy (Burton et al. 2004). More recently an Adenovirus-based trial has failed. In this recent trial, CD8+ lymphocytes were induced against the Gag, Pol, and Nef proteins. Volunteers mounted epitope-specific responses against the candidate immunogens. It is possible that several of these responses were irrelevant since the challenge virus likely differed by 10%. Many of the amino acid replacements in the challenge viruses will have been selected for by CD8+ lymphocytes. Furthermore, the vaccinees probably mounted only three immunodominant responses to only a few epitopes, suppressing development of broader responses to subdominant epitopes. We, therefore, need to focus the best CD8 responses against conserved epitopes and overcome immunodominance issues. The role of CD8+ lymphocytes in control of the AIDS virus and escape CD8+ lymphocytes were first implicated in suppressing HIV replication in 1994 in two studies demonstrating that the reduction in viremia in acute infection was temporally associated with the appearance of HIV-specific CD8+ lymphocytes (Koup et al. 1994, Borrow et al. 1994). A vigorous antibody response occurs subsequent to this initial CD8+ lymphocyte response, after viremia has been controlled. The important role of CD8+ cells was further suggested by work in the simian immunodeficiency virus (SIV)-macaque model of HIV infection. In these studies, anti-CD8 mononclonal antibodies were used to deplete circulating CD8+ lymphocytes. Depletion of circulating CD8+ cells resulted in the loss of control of viremia in both the acute and chronic phases (Matano et al. 1998, Schmitz et al. 1999, Jin et al. 1999, Friedrich et al. 2007). The role of CD8+ lymphocyte escape - that is, viral sequence variation resulting in diminution of recognition by CD8+ lymphocytes - in HIV and SIV pathogenesis is now clear. While recent studies indicate the substantial extent of CD8+ lymphocyte escape in immunodeficiency virus infection, the full extent of escape is unknown and is still, perhaps, underappreciated. Data from the SIV-macaque model have conclusively shown that CD8+ lymphocyte escape occurs in the chronic phase of infection (Evans et al. 1999, 2000). Escape has also been documented during the acute phase (Allen et al. 2000, O'Connor et al. 2002) and throughout the chronic phase (Peyerl et al. 2003, Barouch et al. 2003, Friedrich et al. 2004b, O'Connor et al. 2004) of SIV infection. An analysis of HIV RT sequences in a cohort of > 300 infected individuals demonstrated widespread associations between viral amino acid sequence polymorphisms at certain residues and the expression of particular HLA class I molecules (Moore et al. 2002). It has now been shown that CD8+ T lymphocytes are a widespread and major driving force of SIV (O'Connor et al. 2004) and HIV (Allen et al. 2005) sequence diversity. The cost of CD8+ lymphocyte escape to the virus Recently the fitness cost to the virus of CD8+ lymphocyte escape variants associated with effective control of viremia has been described in both the SIV-macaque model and in HIV infection. The Mamu-A*01-CM9 escape mutation (at position 2 in the epitope) appears to arise only in the presence of putative compensatory mutations (Peyerl et al. 2003, Friedrich et al. 2004b). Attempts to make cloned viruses with only the escape mutation in the epitope have failed (data not shown); mutant viruses are only viable when an additional two mutations are engineered into the CM9-escaped virus. This suggests that these substitutions compensate for a loss of fitness incurred by substitutions in the epitope. Thus, escape mutations may exact a cost to viral fitness. We constructed a variant SIV that bore three escape mutations (Friedrich et al. 2004a). One of these was engineered into Tat (epitope "SL8"), in which escape arises early in the course of infection in Mamu-A*01 positive animals. Another was engineered into Nef ("IW9"), in which escape is detected after an intermediate time in Mamu-B*17-positive animals. The third was in the Mamu-A*01-CM9 epitope, which escapes late. Like Mamu-A*01 (Pal et al. 2002, Zhang 2002, Muhl et al. 2002, Mothe et al. 2003), Mamu-B*17 is an MHC class I allele associated with effective control of SIV (O'Connor et al. 2003, Yant et al. 2006). When this triple-mutant virus was used to infect Mamu-A*01, -B*17-positive animals, the escape mutations were preserved (Friedrich et al. 2004a). However, following infection of Mamu-A*01, -B*17-negative animals with the triple-mutant virus, the CM9 mutation reverted rapidly to wild type, followed by the Nef IW9 mutation. The Tat SL8 mutation, by contrast, persisted for as long as the animals were alive. These data suggest that there is a minimal cost to viral fitness when the Tat SL8 epitope escapes. Complementary studies in HIV infection focused on HLA-B57 and -B5801-positive individuals, because these alleles have been associated with successful containment of HIV infection (Migueles et al. 2000, 2003, Altfeld et al. 2003, 2006, Alabi et al. 2003, Fang et al. 2004, Streeck et al. 2007). The dominant CD8+ lymphocyte specificity in acute infection in HLA-B57-positive subjects is toward the Gag epitope TW10. Characteristically, escape occurs early at position three in the epitope and/or at position nine. Transmission of these escape mutants to HLA-B57/5801-negative subjects resulted in reversion of the mutant to wild type at position three but persistence of the position nine variant, again suggesting that a cost to viral fitness is associated with some escape mutations (Leslie et al. 2004). A spectrum of CD8+ lymphocyte responses may exist that, at one extreme, impose no selection pressure on the virus and at the other extreme, impose intense selection pressure. The Tat-SL8-specific response may be efficacious, but escape occurs at little cost to the virus. The Gag CM9-specific response may be intermediate in its intensity of selection but escape exacts a cost to fitness. A vaccine needs to reduce transmission. The only long-term solution to the HIV epidemic in the developing world is a vaccine that either prevents infection or substantially reduces transmission. The risk of transmission is greatest at the times of highest viremia, that is, in acute infection and in uncontrolled chronic infection. An HIV vaccine, therefore, should aim to limit peak viremia in acute infection and to reduce chronic-phase viral loads from the present median level of ~ 30,000 copies/ml in untreated subjects, to levels at which transmission is unlikely. Infected individuals with viral loads of < 1,700 copies/ml failed to transmit the virus to their HIV-negative partners (Fig. 1) (Quinn et al. 2000, Gray et al. 2001, 2003). The WITS Study in 1999 (Garcia et al. 1999) of over 550 mother-child pairs and found that the risk of mother-to-child transmission was 0% where maternal viral load was < 1,000. Vaccine-mediated reduction in viral load in chronically infected subjects to these levels would reduce transmission in addition to ameliorating the disease course. It will, therefore, be critical to define protective cellular immune responses against the AIDS virus and then to engender these responses by vaccination. Conventional vaccines have not been successful against SIV or HIV challenge Unfortunately, few CD8+ lymphocyte-based vaccine regimens have significantly lowered viral load or affected disease course in macaques using the most stringent SIV challenge models available. Recently several vaccines have claimed success in control of the chimeric SHIV89.6P (Barouch et al. 2000, Amara et al. 2001, Rose et al. 2001, Shiver et al. 2002). However, all but one of those vaccine regimes includes a homologous Env in the vaccine, and serious doubts have surfaced as to the suitability of this virus as a challenge (Feinberg & Moore 2002). The only regimen that did not use Env, a DNA prime/rAdenovirus (rAd) boost encoding only Gag, was ineffective against a SIVmac239 challenge despite its success against SHIV89.6P (Casimiro et al. 2005, McDermott et al. 2005). It is possible, however, that high dose SIV challenge is too stringent a test for our vaccine regimens. The failure of the VAXGEN vaccine trials demonstrates that it will be difficult to protect individuals from HIV challenge with antibody-based vaccines. The infection of 28 volunteers vaccinated with canarypox shows that it will also be difficult for vaccines that induce HIV-specific CD8 responses to protect against HIV challenge (Lee et al. 2004). The recent report of HIV infection of an HLA-B27-positive canarypox vaccinee who made a robust immunodominant response to the Gag KK10 epitope further suggests that current CD8+ lymphocyte-based vaccine modalities are not going to be effective (Betts et al 2005). Finally, the failure of the Merck trial underscores the difficulty of making an effective vaccine for HIV (NPG 2007, Merck 2007). As a result, there is little hope that there will be an effective vaccine for HIV in the near future. Novel vaccine approaches are, therefore, urgently required. There is no " neutralization assay " for CD8+ lymphocytes The correlates of protective immunity against HIV remain a mystery. It was appreciated early on during the AIDS epidemic that patients mounted a broad antibody response to Env. Only a few of these antibodies blocked infection of the virus. Additionally, these neutralizing antibodies were directed against poorly immunogenic regions of the virus. There is a tendency to consider that all CD8+ lymphocyte responses are equivalent, perhaps in the same way that the early Env-specific antibodies were once thought to all be equally effective. It is possible that some CD8+ lymphocyte responses are more efficacious than others. The most effective CD8+ lymphocytes may be the CD8+ lymphocyte analogue of neutralizing antibodies. To resolve this issue, we need to identify which of the many different CD8+ lymphocyte responses present during the acute phase of HIV infection actually contribute to reducing viral replication (Yang 2003). To do this, new sensitive methods of assessing CD8+ lymphocyte function will have to be developed. We have previously relied on autologous EBV-transformed B cell lines pulsed with high concentrations of peptides to assess CD8+ lymphocyte function. Similarly, the popular ELISPOT or ICS assays assess the ability of CD8+ cells to express IFN gamma (IFNg) in response to high concentrations of peptides. None of these assays approximate the ability of CD8+ lymphocytes to suppress the replication of the virus in autologous CD4+ lymphocytes. The various viral proteins are expressed at different times and in different quantities during the viral life cycle (Pomerantz et al. 1990, Klotman et al. 1991, Ranki et al. 1994). Furthermore, the expression of Nef may interfere with CD8+ lymphocyte recognition by down-regulating MHC class I (Collins et al. 1998, Ali et al. 2003, 2004, Collins 2003, Kasper & Collins 2003). More recently, assays investigating the ability of CD8+ lymphocytes to suppress viral replication in cell lines and autologous CD4+ PBMC populations have been developed (Yang et al. 1997, Van Baalen et al. 1998, 2002, Ali et al. 2003, 2004, Loffredo et al. 2005, Tomiyama et al. 2005, Chung et al. 2007). These new assays should help us determine which of the many CD8+ lymphocyte responses can actually control viral replication. Vaccines should target conserved regions of the virus Our current understanding of escape suggests that vaccines may be most effective if they focus efficacious CD8+ lymphocyte responses on selected epitopes that lie in regions of the virus that are under functional or structural constraints. Variation in such regions is unlikely to be well tolerated, since it would be detrimental to viral fitness. As a result, CD8 epitope sequences would likely be preserved, or else accumulate mutations that adversely affect viral fitness. Targeting immune responses toward such regions has the additional benefit of minimizing the sequence differences between vaccine immunogens and circulating HIV strains. CD8+ lymphocyte responses against early-expressed viral proteins may be particularly effective Various investigators have suggested that CD8+ lymphocytes against proteins expressed early in the viral life cycle may be particularly efficacious. CD8+ lymphocyte clones against Nef (Yang et al. 2003a, b) and Rev (Van Baalen et al. 1998, 2002) suppress viral replication in vitro. Indeed, movement of an RT-derived CD8+ lymphocyte epitope from Pol to Nef resulted in increased susceptibility of cells infected with the recombinant virus to lysis by an RT-specific CD8+ lymphocyte clone (Van Baalen et al. 2002). These data and early vaccine work (Gallimore et al. 1995) suggest that CD8+ lymphocytes against these early proteins play an important role in containment of viral replication. Unfortunately, many of these experiments have relied on a limited number of well-defined clones, and the general applicability of these findings remains to be determined. Vaccines must focus immune responses rather than relying on natural immunodominance patterns It is becoming increasingly apparent that not all CD8+ lymphocytes are created equal. It is likely that inducing the CD8+ lymphocyte responses that are present in natural infection will not be in every vaccinee's best interests. The aim of a vaccine should not be simply to induce as many CD8+ lymphocyte responses as possible. In many cases, it will be necessary to alter the natural immunodominance of the HIV- or SIV-specific CD8+ lymphocyte response since immunodominant responses may limit the development of potentially more effective sub-dominant responses (Chen et al. 2000, Palmowski et al. 2002, Rodriguez et al. 2002, Smith et al. 2005, Yewdell 2006). For a vaccine, it may be important to force escape at a price to the virus. For AIDS vaccine development, it will be important to define efficacious CD8+ lymphocyte responses against HIV and SIV. These CD8+ lymphocytes should be directed against epitopes that:
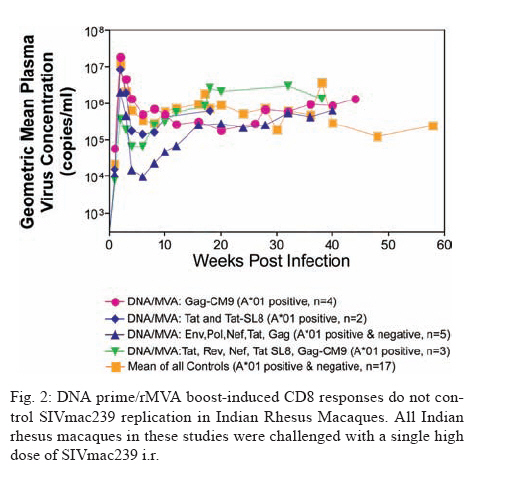
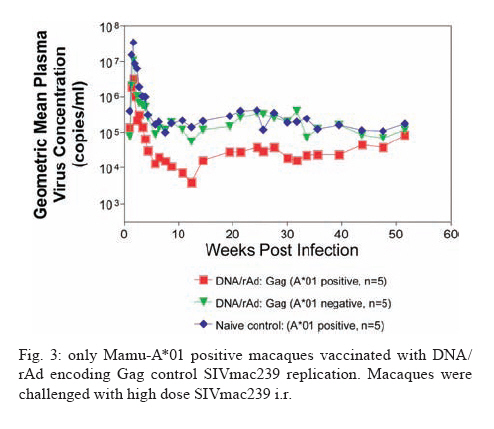
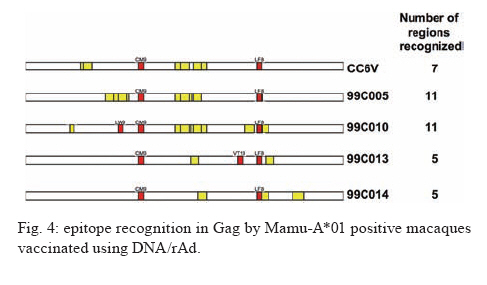
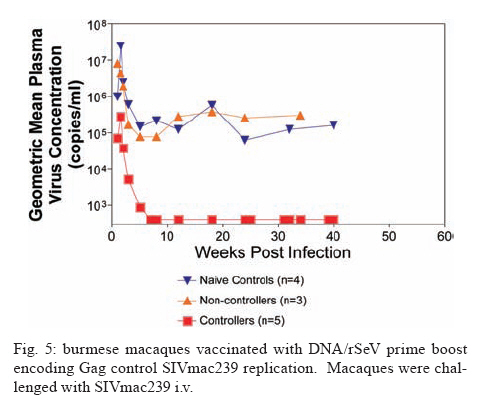
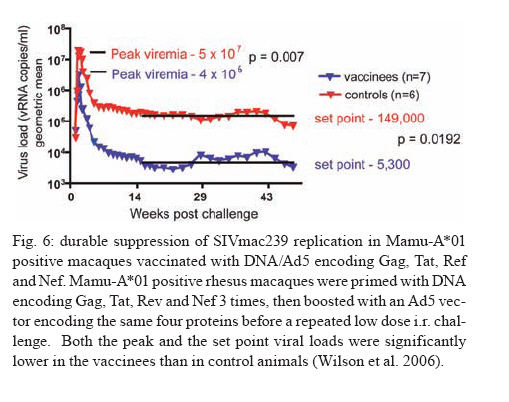
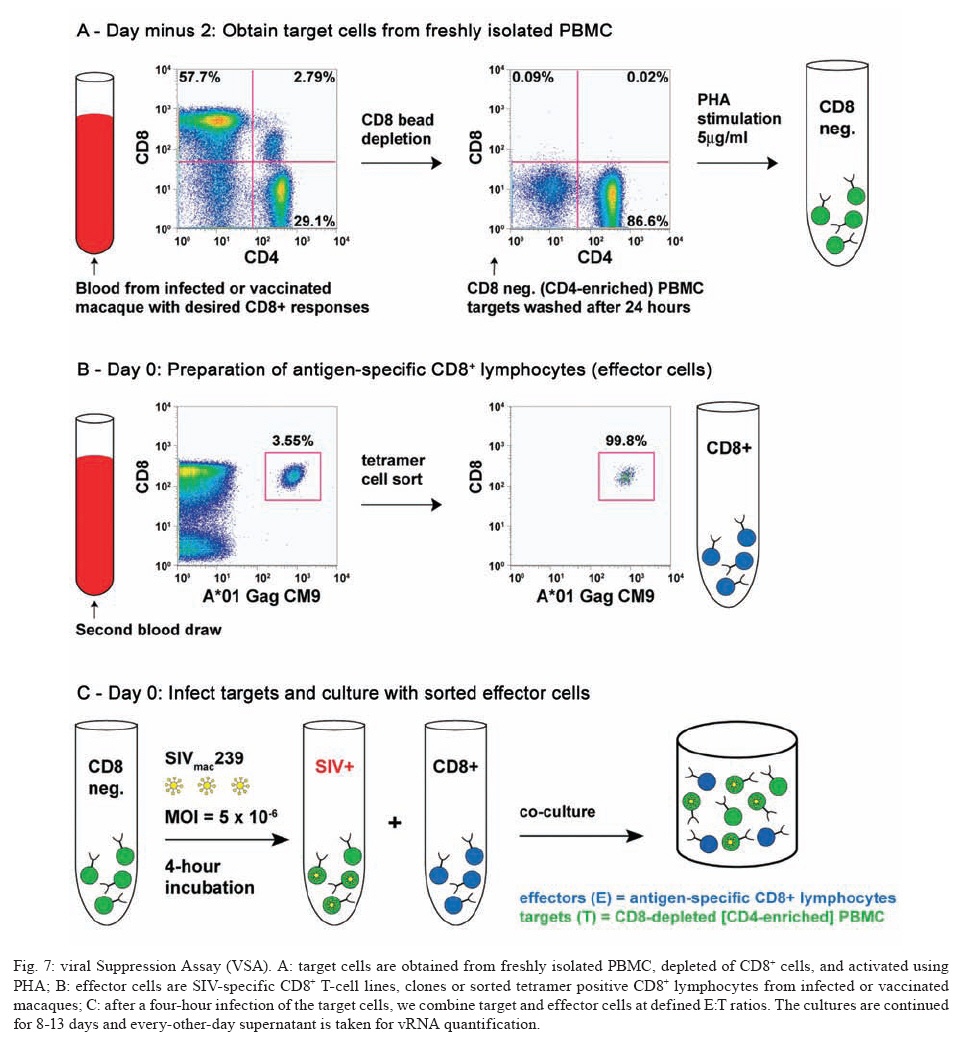
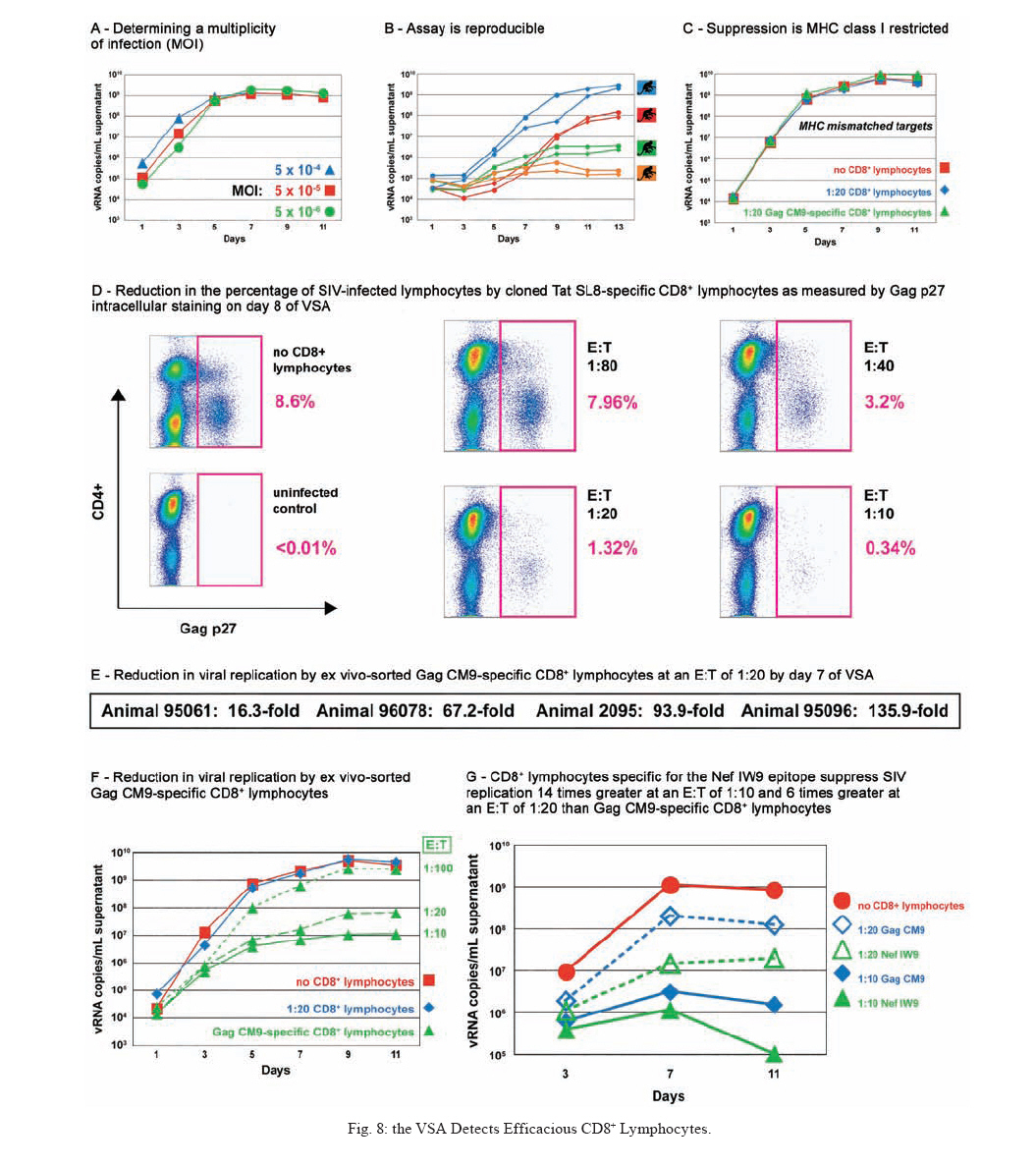
REVIEW OF NON-HUMAN PRIMATES SIV CHALLENGE STUDIES Effects of Vaccine-Induced CD8+ lymphocytes on the Course of SIVmac239 Infection Vaccine-induced CD8+ lymphocytes have been implicated in the control of virus replication in SIV-challenged and SHIV89.6P-challenged macaques. Therefore, we wanted to test the impact that vaccine-induced CD8+ lymphocyte responses against an immunodominant Gag epitope might have in the absence of other immune responses. By themselves, these strong CD8+ lymphocyte responses failed to control SIVmac239 replication (Allen et al. 2002a) (Fig. 2). The regulatory proteins of HIV may represent important vaccine targets. We then assessed the role of Tat-specific CD8+ lymphocytes in controlling pathogenic SIVmac239 replication after using a DNA prime/rMVA boost vaccine regimen. Despite the induction of Tat-specific CD8+ lymphocytes, there was no significant reduction in either peak or viral set point compared to that of controls (Allen et al. 2002b) (Fig. 2). We next used a DNA prime/rMVA boost regimen to immunize rhesus macaques against nearly all SIV proteins. These animals were also challenged with SIVmac239. The immunization regimen resulted in the induction of virus-specific CD8+ and CD4+ responses in all vaccinees. Vaccinated animals had reduced peak viremia compared with controls. However, despite the induction of virus-specific cellular immune responses and reduced peak viral loads, most animals still suffered from gradual CD4 depletion and progressed to disease (Horton et al. 2002) (Fig. 2). We then vaccinated Mamu-A*01+ macaques with constructs encoding a combination of CD8+ lymphocyte epitopes and full-length proteins (Tat, Rev, and Nef) by using a DNA prime/rMVA boost regimen. The vaccination induced virus-specific CD8+ lymphocytes and CD4+ helper T lymphocytes with CD8+ lymphocyte frequencies as high as 20,000/106 PBMC. The final rMVA vaccination, delivered intravenously, engendered long-lived mucosal CD8+ lymphocytes. At 16 weeks after the final rMVA vaccination, the vaccinees and naive Mamu-A*01+ controls were challenged i.r. with SIVmac239. Massive early anamnestic cellular immune responses controlled acute-phase viral replication; however, the three vaccinees were unable to control virus replication in the chronic phase. This study suggested that multispecific mucosal CD8+ lymphocytes, in the absence of neutralizing antibodies, can achieve a modicum of control over early viral replication, but they are unable to control chronic-phase viral replication after a high-dose mucosal challenge with a pathogenic SIV (Vogel et al. 2003) (Fig. 2). Previous studies (Matano et al. 2004) showed that macaques vaccinated with DNA prime/rAd boost encoding SIV Gag controlled replication of SHIV89.6P. To test this vaccination regimen in a more stringent challenge model, macaques were either vaccinated with a DNA prime/ rAd boost or a rAd prime and a rAd boost. Only Mamu-A*01-positive macaques vaccinated using the DNA prime/rAd boost controlled viral replication (Fig. 3). Vaccination with rAd/rAd was ineffective in both Mamu-A*01-positive and -A*01-negative macaques, and Mamu-A*01-negative animals vaccinated with DNA prime/rAd boost failed to control SIVmac239 replication. We determined the breadth of the immune response in the Mamu-A*01-positive macaques that controlled viral replication. They all made similar epitope-specific immune responses to Mamu-A*01-restricted epitopes in Gag (Fig. 4). However, more than two-thirds of the responses against the vaccine construct were directed against the immunodominant Gag CM9 epitope. Since only DNA-primed/rAd-boosted Mamu-A*01+ macaques control viral replication, it is likely that Mamu-A*01-restricted CD8+ responses against epitopes in Gag are at least partly efficacious against SIV. Control of highly pathogenic SIVmac239 replication by vaccine-induced CD8+ lymphocytes To date no vaccine regimen has been successful in the containment of replication of the pathogenic SIV that induces chronic disease progression. Indeed, it has remained unclear if vaccine-induced CD8+ lymphocytes can control SIV replication. Eight Burmese rhesus macaques vaccinated with DNA-prime/Gag-expressing recombinant Sendai (rSeV) virus vector-boost were challenged intravenously with SIVmac239. Five of the vaccinees controlled viral replication and had undetectable plasma viremia after five weeks of infection (Matano et al. 2004) (Fig. 5). CD8+ lymphocytes from all five of these macaques rapidly selected for escape mutations in Gag, indicating that vaccine-induced CD8+ lymphocytes successfully contained replication of the challenge virus. Interestingly, analysis of the escape variant selected in three vaccinees that share a MHC class I haplotype revealed that the escape variant virus was at a replicative disadvantage compared to SIVmac239. These findings suggested that the vaccine-induced CD8+ lymphocytes had " crippled " the challenge virus. These results indicate that vaccine induction of effective CD8+ lymphocytes by DNA/prime followed by Sendai virus boost can result in the containment of replication of a highly pathogenic immunodeficiency virus if those CD8+ lymphocytes target constrained epitopes. DNA priming followed by Ad5 (Gag, Tat, Rev and Nef) boosting in Mamu-A*01 positive macaques controls replication of SIVmac239. Whether vaccine-induced cellular immunity in the absence of any Env-specific antibodies can control viral replication was studied by using multiple low-dose challenges with the highly pathogenic SIVmac239 isolate (Wilson et al. 2006). In this experiment, eight Mamu-A*01 positive Indian rhesus macaques were vaccinated with SIV Gag, Tat, Rev and Nef using a DNA prime, adenovirus boost strategy. Peak viremia (p = 0.007) and the chronic phase, set point viral load (p = 0.0192) were significantly decreased in the vaccinated cohort, out to one-year post infection (Fig. 6). The loss of CD4+ T-cell memory populations was also reduced in the vaccinated animals. Of note is that only one of the eight vaccinees had developed Env-specific neutralizing antibodies by one year after infection. Overall, the control of infection was a significant improvement over that observed in animals vaccinated with SIV Gag only. However, it should be noted that these macaques were challenged intrarectally with repeated low doses of SIVmac239, whereas a single high dose intrarectal challenge was used in the studies of the DNA/Ad5 Gag vaccine (Casimiro et al. 2005, McDermott et al. 2005). Whether the different challenge regimen or the alteration in the vaccine composition (Gag alone, rather than Gag, Tat, Rev and Nef) is responsible for the different outcome is unknown, but is now testable. Nonetheless, vaccine-induced cellular immune responses can clearly exert a measure of control over replication of a primate immunodeficiency virus in the complete absence of neutralizing antibodies. This finding provides some grounds to believe that a vaccine designed to induce only cellular immune responses might be able to control viral replication. New assays for CD8+ lymphocyte function It has been difficult to study CD8+ lymphocyte function in vitro. Recently, several groups have shown that in vitro-expanded CTL suppress HIV replication in cell lines and primary cells (Yang et al. 1997, Ali et al. 2003, 2004, Tomiyama et al. 2005, Van Baalen 1998, 2002). We have developed a similar viral suppression assay (VSA) in macaques (Loffredo et al. 2005, 2007, Chung et al. 2007) (Fig. 7). In the VSA, CD8+ lymphocytes of the desired specificity are tetramer-sorted directly ex vivo grown in cell lines or cloned and are added to acutely infected autologous CD4+ targets (Fig. 7). We can then assess the ability of the sorted, peptide-specific CD8+ lymphocytes to suppress virus replication by measuring the accumulation of viral RNA genomes in the culture supernatant using sensitive quantitative polymerase chain reaction (QPCR). We already have shown that a multiplicity of infection (MOI) of 5 x 10-6 is optimal for infecting targets (Fig. 8A). We have shown that viral replication is consistent from culture to culture (Fig. 8B) and that suppression of viral replication is MHC class I restricted (Fig. 8C). Recently, we have used intracellular p27 staining to monitor infection of target cells (Fig. 8D). As shown inFig. 8D, addition of cloned Tat SL8 specific CD8+ lymphocytes reduce infection of CD4+ lymphocytes at day 8 post infection as assessed by intracellular p27 staining. We have already used the VSA to test some CD8+ lymphocytes that are present at high frequencies in vivo. For example, Gag CM9-specific CD8+ lymphocytes (freshly sorted from PBMC) reduce SIVmac239 replication at least ten-fold when added to cultures (Fig. 8E-F). Interestingly, Nef IW9-specific CD8+ lymphocytes are especially efficient at suppressing viral replication (Fig. 8G). Repeated low-dose mucosal SIVmac239 challenge results in the same viral and immunological kinetics as high-dose challenge: a model for the evaluation of vaccine efficacy in nonhuman primates SIV challenge of rhesus macaques provides a relevant model for the assessment of HIV vaccine strategies. To ensure that all macaques become infected, the vaccinees and controls are normally exposed to large doses of pathogenic SIV. These nonphysiological high-dose challenges may adversely affect vaccine evaluation by overwhelming potentially efficacious vaccine responses. To determine whether a more physiologically relevant low-dose challenge can initiate infection and cause disease in Indian rhesus macaques, we used a repeated low-dose challenge strategy designed to reduce the viral inoculum to more physiologically relevant doses. In an attempt to more closely mimic challenge with HIV, we administered repeated mucosal challenges with 30, 300, and 3,000 50% tissue culture infectious doses (TCID50) of pathogenic SIVmac239 to six animals in three groups. Infection was assessed by sensitive quantitative reverse transcription-PCR and was achieved following a mean of 8, 5.5, and 1 challenge(s) in the 30, 300, and 3,000 TCID50 groups, respectively. Mortality, humoral immune responses, and peak plasma viral kinetics were similar in five of six animals, regardless of challenge dose. Interestingly, macaques challenged with lower doses of SIVmac239 developed broad T-cell immune responses as assessed by ELISPOT assay. This low-dose repeated challenge may be a valuable tool in the evaluation of potential vaccine regimens and offers a more physiologically relevant approach for pathogenic SIVmac239 challenge experiments (McDermott et al. 2004). CONCLUSIONS We have shown that DNA prime/rAd boost encoding Gag reduces viral replication in SIVmac239-challenged Mamu-A*01-positive Indian macaques (Casimiro et al. 2005, McDermott et al. 2005). The same regimen in Mamu-A*01-negative macaques fails to control replication, implicating Mamu-A*01-restricted CD8+ lymphocytes in this control. Similarly a DNA prime/rSeV boost encoding Gag reduces viral replication in SIVmac239-challenged Burmese macaques expressing particular MHC class I molecules (Matano et al. 2004). Finally, the recent results indicating that a DNA prime Ad5 boost vaccine encoding Gag, Tat, Rev, and Nef can control SIVmac239 replication give us hope that a CTL-based vaccine is possible (Wilson et al 2006). This suggests that it is possible to blunt replication of this highly pathogenic SIV, if CD8+ lymphocyte responses are induced against the appropriate epitopes. We and others have now developed a viral suppression assay (VSA) to determine which of the many CD8+ lymphocyte responses are the most efficient at controlling viral replication (Yang et al. 1997, Van Baalen et al. 1998, 2002, Ali et al. 2003, 2004, Loffredo et al. 2005, Chung et al. 2007, Tomiyama et al. 2005). We now need to define epitopes in regions of the virus that are targeted by effective CD8+ lymphocytes and cannot tolerate variation. We also need to design vaccine strategies to test whether vaccine-induced CD8+ lymphocyte responses against these epitopes can reduce viral replication in vivo. We should attempt to reduce the viral set point by two logs; this should reduce transmission of the virus and increase longevity of the infected vaccinee. Attempts to develop vaccines that induce broadly neutralizing antibodies have failed. Few CTL-inducing vaccine regimens have proved to be useful against SIV. Novel, innovative vaccine design, therefore, remains a priority. We need to define efficacious CD8+ lymphocyte responses using novel assays. We need to induce these efficacious CD8+ responses using CD8+ epitopes expressed by single naked DNA and viral vectors to overcome immunodominance problems. We therefore need an entirely new, innovative vaccine regimen against novel targets. To John Loffredo and Amanda Espinosa for help in preparing this article. REFERENCES
Copyright 2008 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc08022f1.jpg] [oc08022f4.jpg] [oc08022f7.jpg] [oc08022f2.jpg] [oc08022f5.jpg] [oc08022f8.jpg] [oc08022f3.jpg] [oc08022f6.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}