 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 104, No. 2, March, 2009, pp. 162- 169 Host genetic and epigenetic factors in toxoplasmosis Sarra E JamiesonI, II; Heather CordellII, III; Eskild PetersenIV; Rima McLeodV, VI; Ruth E GilbertVII;Jenefer M BlackwellI, II, + ITelethon
Institute for Child Health Research, Centre for Child Health Research, The University
of Western Australia, PO Box 855, West Perth, Western Australia 6872, Australia Received 10 October
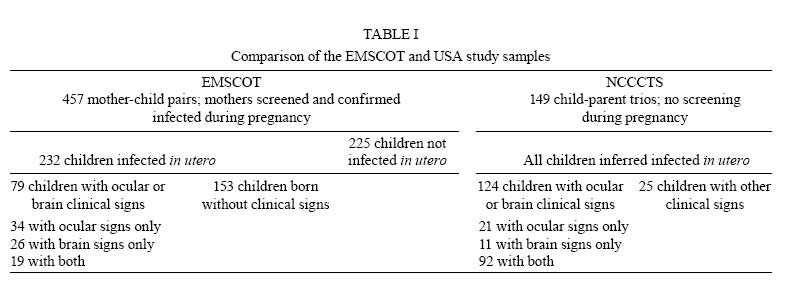
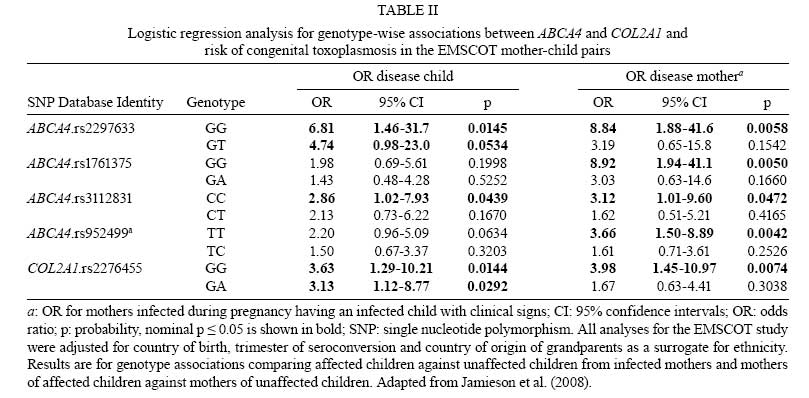
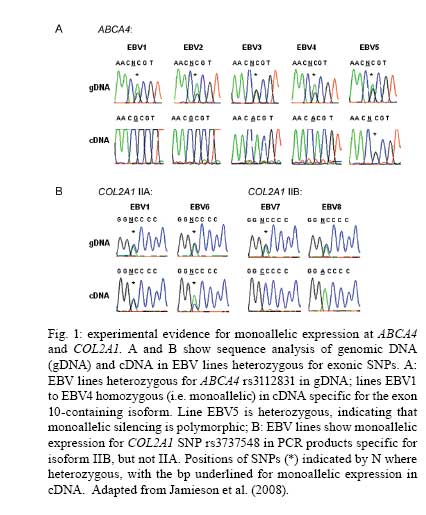
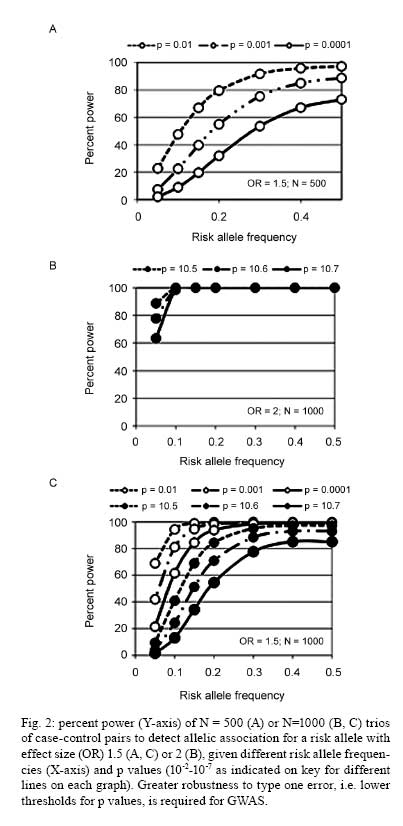
2008 Financial support: Main funders for the published work on which this article is based were Guide Dogs for the Blind Association, UK; NIH, USA. Code Number: oc09029 ABSTRACT Analysing human genetic variation provides a powerful tool in understanding risk factors for disease. Toxoplasma gondii acquired by the mother can be transmitted to the fetus. Infants with the most severe clinical signs in brain and eye are those infected early in pregnancy when fetal immunity is least well developed. Genetic analysis could provide unique insight into events in utero that are otherwise difficult to determine. We tested the hypothesis that propensity for T. gondii to cause eye disease is associated with genes previously implicated in congenital or juvenile onset ocular disease. Using mother-child pairs from Europe (EMSCOT) and child/parent trios from North America (NCCCTS), we demonstrated that ocular and brain disease in congenital toxoplasmosis associate with polymorphisms in ABCA4 encoding ATP-binding cassette transporter, subfamily A, member 4 previously associated with juvenile onset retinal dystrophies including Stargardt's disease. Polymorphisms at COL2A1 encoding type II collagen, previously associated with Stickler syndrome, associated only with ocular disease in congenital toxoplasmosis. Experimental studies showed that both ABCA4 and COL2A1 show isoform-specific epigenetic modifications consistent with imprinting, which provided an explanation for the patterns of inheritance observed. These genetic and epigenetic risk factors provide unique insight into molecular pathways in the pathogenesis of disease. Key words:congenital infection - toxoplasmosis - genetics - epigenetics Following the human genome project, analyzing human genetic variation has emerged as a powerful tool in understanding both direct genetic and modifiable environmental (Davey Smith et al. 2008, Ebrahim & Davey Smith 2008) risk factors for disease. These approaches could be applied to analysis of clinical outcomes for both congenital and acquired toxoplasmosis. We recently used a genetic approach to look at congenital toxoplasmosis (Jamieson et al. 2008). As is well known, Toxoplasma gondii is a ubiquitous protozoan parasitic infection that, if acquired for the first time during pregnancy, can be transmitted to the fetus. At birth, infants infected in utero may have intracranial calcification, hydrocephalus and ocular disease broadly defined as retinochoroiditis or inflammation of the retina and choroid with associated vitritis (Gilbert & Gras 2003, Gras et al. 2005, McLeod et al. 2006). New ocular lesions can occur at any age after birth, in untreated and some treated children. Whilst severity of disease is influenced by trimester in which infection is acquired by the mother (Dunn et al. 1999, Remington et al. 2005), other factors including genetic predisposition may contribute. For example, previous studies suggest that genes affecting immune response, including HLA (Mack et al. 1999), influence clinical outcome in the child. However, since infants who have the most severe clinical signs in the brain and eye are those infected early in pregnancy (Dunn et al. 1999, Remington et al. 2005) when fetal immunity is least well developed, we considered whether genes that encode molecules that play a role in developmental processes in the eye or the brain could provide another avenue to identify candidate genes that contribute to clinical phenotype observed in the child. This could provide unique insight into events in utero that determine the clinical outcome of infection. The eye disorder gene hypothesis? One hypothesis we pursued (Jamieson et al. 2008) was whether the propensity for T. gondii to cause eye disease was associated with genes previously implicated in congenital or juvenile onset ocular disease. Two genes were studied, ABCA4 encoding ATP-binding cassette transporter subfamily A member 4 associated with juvenile onset retinal dystrophies including Stargardt's disease (Ducroq et al. 2002, Koenekoop 2003), and COL2A1 encoding type II collagen associated with Stickler syndrome (Rose et al. 2005), in which there is congenital abnormal vitreous and lattice retinal degeneration. The full results of these studies are presented elsewhere (Jamieson et al. 2008). All of the data provided here are from this paper, which is therefore not further referenced in the results section below. Here we summarize the salient findings and examine the implications of this research to the application of genetics as a tool to understand the multiple pathologies associated with toxoplasmosis. Cohorts for genetic studies Two unique cohorts (Table I) were available to test this novel candidate gene hypothesis. These cohorts are from the European Multicentre Cohort Study on Congenital Toxoplasmosis (EMSCOT), which recruited prospectively for mothers with primary T. gondii in pregnancy (Gilbert & Gras 2003, Gras et al. 2005), and from the National Collaborative Chicago-based Congenital Toxoplasmosis Study (NCCCTS) (McLeod et al. 2006), in North America, to which infants and children with congenital infection with T. gondii are referred. The EMSCOT cohort - For the EMSCOT cohort, DNA was successfully obtained from 457 mother-child pairs with confirmed infection during pregnancy. Transmission of infection during pregnancy was confirmed in 232 (51%) infants; 225 (49%) infants remained uninfected. All infants were monitored for clinical signs until at least three years of age and many have been followed for > 10 years. Of the 232 infected infants, 79 (34%) had clinical signs (referred to as affected) of congenital toxoplasmosis: 53 (67%) with ocular lesions (retinochoroidal lesions), 45 (57%) with brain lesions (hydrocephalus or intracranial calcifications detected on ultrasound examination of the brain), 19 (24%) of these infants had both eye and brain lesions. One hundred fifty three (66%) infected infants had no clinical signs of disease up to a minimum follow-up period of three years of age. Two-thirds of mother-child pairs reported grandparents' countries of birth. Of these, 95% were Caucasian and 5% of African origin. The NCCCTS cohort - For the second independently ascertained NCCCTS cohort, DNA was successfully obtained from 149 children with confirmed congenital infection (69% Caucasian, 15% Hispanic, 8% Asian or Pacific Islander, 3% African American, 0.7% Native American, 4.7% mixed race) plus available parents. At birth or time of diagnosis, 92 (62%) infected children had brain calcifications with/without hydrocephalus and retinal lesions, 21 (14%) had retinal lesions only, 11 (7%) had brain calcifications with/without hydrocephalus only and 25 (17%) infected children were without these clinical findings. Only the 124 children with confirmed clinical findings in eye and/or brain were included in the allelic association analysis for this cohort. This provided data on 124 trios classified as affected (i.e. eye or brain lesions or both), 113 trios classified as eye disease (with or without brain disease) and 103 trios classified as brain disease (with or without eye disease). Results of genetic analyses Seven single nucleotide polymorphisms (SNPs) at ABCA4 and seven SNPs at COL2A1 (Jamieson et al. 2008) were genotyped in both cohorts. Genetic associations with clinical outcome For the EMSCOT cohort, nominally significant (i.e. p values without correction for multiple testing) allelic associations were observed for SNPs at ABCA4 and at COL2A1 when affected children (i.e. children with retinal or brain disease or both) in the EMSCOT cohort were compared with infected but unaffected children. Large effect sizes (odds ratios or OR 2.9-6.8) were observed for these associations, particularly in children or homozygous for the disease allele (Table II). Significance at both genes was improved when the analysis was enriched for children with the eye lesion phenotype (with/without brain lesions, i.e. leaving out children with brain disease only) compared to unaffected children, but not when the analysis was similarly enriched for children with brain lesions (with/without eye lesions, leaving out children with eye disease only) compared to unaffected children. In the NCCCTS cohort, significant allelic associations were observed at ABCA4 and COL2A1 under a dominant model of inheritance (Table III). The use of case/parent trios and transmission disequilibrium testing in FBAT controlled for ethnic admixture in this cohort. For COL2A1, significance improved when the analysis was enriched for the eye lesion phenotype (with/without brain lesions), with the associated markers all within one strong haplotype block. Overall, the evidence from the children with eye and brain signs associated with congenitally acquired toxoplasmosis in these two cohorts was for association with eye disease and these two previously recognized eye disorder genes, ABCA4 and COL2A1. Influence of mother's genotype on clinical outcome To determining whether mother's genotype had an effect on disease outcome in the child, logistic regression analysis was carried out comparing mothers of affected children (eye and/or brain disease) with mothers of infected unaffected children for the EMSCOT cohort. This proved interesting in that (i) most of the associations were more significant in comparisons of mothers than in comparisons of infants (Table II), again with large effect sizes (OR 3.1-8.9), (ii) additional ABCA4 and COL2A1 were associated with mothers of affected infants compared to mothers of unaffected infants and (iii) all of the ABCA4 SNPs but none of the COL2A1 SNPs were associated with mothers of infants with brain disease suggesting a role for ABCA4 in brain pathology. Statistically, it appeared that genetic effects of these two loci on disease in the child were being diluted out in making direct comparisons of the affected versus unaffected children relative to the evidence for association when comparing the mothers. Unusual patterns of inheritance observed when comparing genotype-wise associations in children versus mothers suggested that there may be a direct effect of mother's genotype, or that there may be parent-of-origin effects (imprinting), i.e. that for the child it is the origin of, and not just the combination of, alleles that is important in determining disease risk. Using a log-linear method previously designed to evaluate maternal genotype and/or parent-of-origin effects in case-parent trios (Weinberg et al. 1998), evidence for imprinting given mother's and child's genotypes was obtained at ABCA4 (p = 0.033) and COL2A1 (p = 0.05) in the NCCCTS trios. For EMSCOT we adapted the method for use with mother-child pairs and found evidence for effects of maternal genotype given child's genotype and imprinting for two SNPs at COL2A1 (p = 0.0088 and p = 6.87 x 10-5) and evidence for imprinting taking account of both mother's and child's genotype for one COL2A1 SNP (p = 0.0025). Given the statistical limitations of small sample size and power in the modeling analysis and the fact that a direct effect of mother's genotype seemed unlikely biologically, we looked for experimental evidence of epigenetic effects or imprinting for ABCA4 and COL2A1. Evidence for imprinting To obtain experimental evidence for imprinting, an anonymised EBV B cell bank was screened (i) to find individuals heterozygous for exonic SNPs at both loci and (ii) to determine mono-allelic expression in elicit transcripts obtained from RNA from these EBV cell lines. EBV lines were identified that were heterozygous for SNP rs3112831 in exon 10 of ABCA4 and for SNP rs3737548 in exon 7 of COL2A1. For the known functional exon 10-containing isoform at ABCA4, four of the five EBV lines that were heterozygous for genomic DNA showed monoallelic expression in cDNA for the exon 10 rs3112831 SNP (Fig. 1A). The most likely explanation for this was polymorphic imprinting of the paternally-derived allele. Monoallelic expression was also observed (Fig. 1B) for a SNP in exon 7 in cDNA specifically amplified for the IIB short form of COL2A1, but not for cDNA amplified for the IIA long form, consistent with isoform-specific imprinting in this case with the maternally-derived allele silenced. DISCUSSION In our published study (Jamieson et al. 2008) we examined the specific genetic hypothesis that polymorphisms in two genes known to be associated with ocular disorders, ABCA4 and COL2A1, are associated with eye disease caused by congenital toxoplasmosis. These associations were replicated across two independently ascertained cohorts, providing further confidence in this result. How these findings are related to the direct functional roles of ABCA4 and COL2A1 are discussed in more detail in the original paper. The observation that both loci were influenced by epigenetic modification of gene expression was also novel and exciting. The study provided new insight into processes that occur in early in embryonic development when it is not easy to determine what is happening when the fetus is first infected with a parasite such as T. gondii. The specific genetic associations observed have seeded follow-up studies to address how the parasite triggers the development of ocular disease during embryogenesis and fetal development. What is known about the chronological expression and localization of COL2A1 and ABCA4 during development? How does the parasite influence this? Since T. gondii is a potent trigger for, and direct regulator of, the NFκB signaling pathway (Molestina & Sinai 2005a, b), could the parasite trigger disease via polymorphic NFκB binding in the promoter regions of these two genes? Does the parasite directly interfere with methylation and/or histone acetylation patterns of host DNA, thereby directly affecting epigenetic regulation of gene expression? Studies in progress are designed to test some of these hypotheses. In this initial genetic study of congenital toxoplasmosis we took a specific hypothesis-driven candidate gene approach. Such an approach could also be applied, for example, to developmental genes previously associated with other clinical outcomes of congenital toxoplasmosis such as hydrocephalus. Alternatively, given a sufficiently large sample size, further studies could take a non-hypothesis-driven genome-wide approach to identify critical developmental pathways and pathogenic processes associated with congenital disease. Since other congenital infections, e.g. cytomegalovirus, often elicit similar pathologies, it would also be of interest to test both this hypothesis-driven candidate gene analysis to determine whether the same or different genes influence specific pathologies (such as eye or brain lesions) and/or genome-wide approaches across cohorts of different congenital and perinatally-acquired infectious diseases. Understanding more about clinical outcomes associated with T. gondii infection acquired in childhood could also benefit from the application of the tools of modern post-genomic genetic and epigenetic analysis. One major problem with all candidate gene studies for infectious diseases reported to date is that they were under-powered (Burgner et al. 2006). Until recently, this was a general problem in genetic analysis of complex disease, along with issues relating to study design and population history (Cordell & Clayton 2005, Palmer & Cardon 2005). Fig. 2 compares power to detect association at OR = 1.5 or OR = 2, given different risk allele frequencies, p values and sample sizes. This shows that 500 trios or case-control pairs have little power to detect association for small effect sizes (OR = 1.5). Even with 1,000 trios or case-control pairs, power is limited for risk alleles with frequency < 0.2 for an effect size (OR) 1.5, although low frequency (e.g., 0.10) risk alleles with larger effect sizes (OR > 2) may be detected. This means that the studies we can perform on congenital toxoplasmosis using the current EMSCOT and NCCCTS resources are only powered to detect large effect sizes (OR > 2). To carry out further candidate gene studies of congenital or acquired toxoplasmosis with confidence, the research community needs to work together to develop much larger cohorts for primary and replication studies. This applies also to the application of SNP-chip based genome-wide association studies which have proved to be quite successful in rapidly increasing the number of loci that have been positively associated with complex diseases. For example, the Consortium WTCCC study (2007) of 14,000 cases of seven common diseases and 3,000 shared controls has itself identified 24 independent association signals at p < 5 x 10-7, nine of which were in Crohn's disease, three in rheumatoid arthritis, seven in type 1 diabetes and three in type 2 diabetes. The increased problem of multiple testing associated with genotyping in excess of 500,000 SNPs in large numbers of cases and controls necessitated this stringent threshold p value < 5 x 10-7 to achieve genome-wide significance. However, across all diseases, a large number of further signals, including 58 loci with single point p values between 10-5-5 x 10-7, were identified which are likely to yield additional susceptibility loci. A number of papers providing validation of the original WTCCC data have already been published (Cooper et al. 2007, Parkes et al. 2007, Plenge et al. 2007, Samani et al. 2007, Thomson et al. 2007, Todd et al. 2007, Zeggini et al. 2007, Beckly et al. 2008), including one on type 1 diabetes that increases the number of loci with compelling evidence of association from six to at least 10 (Todd et al. 2007). The WTCCC study is but one of an increasing number of published GWAS for complex diseases (Buch et al. 2007, Burton et al. 2007, Cupples et al. 2007, Duggan et al. 2007, Easton et al. 2007, Florez et al. 2007, Gudmundsson et al. 2007, Hampe et al. 2007, Kathiresan et al. 2007, Raelson et al. 2007, Roberts et al. 2007, Scuteri et al. 2007, Sladek et al. 2007, Sulem et al. 2007, Tomlinson et al. 2007, Zanke et al. 2007, Cronin et al. 2008). As pointed out in the recent News Feature on "Genetics by Numbers" in Nature (Baker 2008), identification of the initial SNP association on a primary GWAS is only the first step on the path to validating and identifying the etiological genes and associated mechanisms of disease susceptibility. Some papers have demonstrated at least two independent etiological variants at one locus (6q23) associated with rheumatoid arthritis, while regions like the HLA complex require intensive fine mapping. For example, following the WTCCC type 1 diabetes study, a combined total of 1,729 polymorphisms across HLA were genotyped in > 6000 cases and controls across cohorts and statistical methods of recursive partitioning and regression applied to pinpoint disease susceptibility to the MHC class I genes HLA-B and HLA-A [risk ratios > 1.5; p (combined) = 2.01 x 10-19 and 2.35 x 10-13, respectively] in addition to the established associations of the MHC class II HLA-DQB1 and HLA-DRB1 genes (Nejentsev et al. 2007). This demonstrates the intensive mapping that must follow any primary GWAS to validate the SNP associations and identify the genes associated with disease and the statistical power that can be achieved by genotyping large numbers of cases across multiple cohorts. The application of GWAS technology to the study of complex disease has also stimulated development of genetic statistical tools, with recent papers highlighting the use of localized haplotype cluster analysis to improve detection of disease associated variants (Browning & Brownin 2008), better methods for correcting for population stratification by identifying hidden population structures (Li & Yu 2008), robust methods to allow primary screening and replication using the same data set in family-based association testing (Van Steen et al. 2005), a new multipoint method for GWAS by imputation of genotypes based on knowledge of tag-SNPs (Marchini et al. 2007) and the effect of local LD patterns around the primary analysis signal(s) and tagging efficacy of typed markers in determining a suitable density of tag-SNPs for fine-mapping (Wiltshire et al. 2008). All of these have implications for study design, which given the global distribution of toxoplasmosis, need to be carefully planned. Recent papers discuss many of the issues that affect modern genome-wide approaches to genetic association studies using case-control and trio designs (Teo 2008, Teo et al. 2008a, b, 2009a, b). Case-control approaches are more efficient in terms of the amount of genotyping required, but will not permit detection of maternal genotype effect, maternal-foetal interactions or parent-of-origin (imprinting) effects (Buyske 2008). Family-based studies (minimally mother-child pairs, or child/parent trios) make it possible to distinguish between these different mechanisms (Weinberg et al. 1998, Cordell et al. 2004, Hsieh et al. 2006) and are also valuable in controlling for mixed ethnicity or ethnic admixture in the population under study. Our specific study of congenital toxoplasmosis has demonstrated how important these effects can be. It is also of note that, despite the exciting results of highly-powered genome-wide case-control association studies, the genes identified to date generally fail to account for all of the attributable genetic risk. This suggests that many genes are being missed by failure to take these other models and the huge role that epigenetic effects are likely to play, into account. In summary, we believe that the application of a genetic approach has provided novel insight into disease mechanisms in congenital toxoplasmosis. We hope this will seed new collaborative consortia that will allow application of this technology with greater efficiency and with adequate statistical power to determine associations of relevance to our understanding of the underlying mechanisms of disease. ACKNOWLEDGMENTS To the patients, their families and their physicians, for their participation in EMSCOT and the NCCCTS. REFERENCES
Copyright 2009 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc09029t3.jpg] [oc09029t1.jpg] [oc09029f1.jpg] [oc09029t2.jpg] [oc09029f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}