 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 104, No. 4, July, 2009, pp. 531-548 REVIEW Revisiting steroid treatment for septic shock: molecular actions and clinical effects - a review André M JapiassúI, II, IV; Jorge IF SalluhI, III; Patrícia T BozzaI; Fernando A BozzaII; Hugo C Castro-Faria-NetoI, + ILaboratório
de Imunofarmacologia, Instituto Oswaldo Cruz Financial support: PAPES, FAPERJ, CNPq Received 15 April
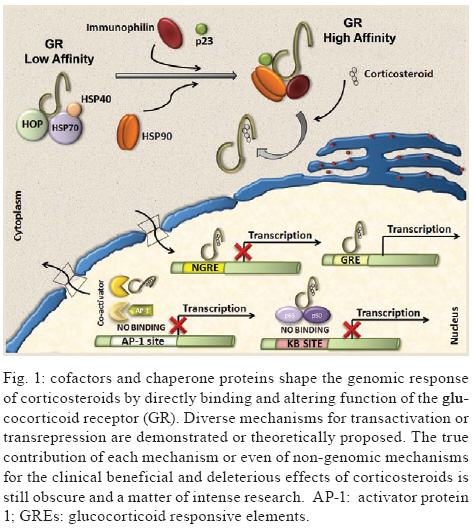
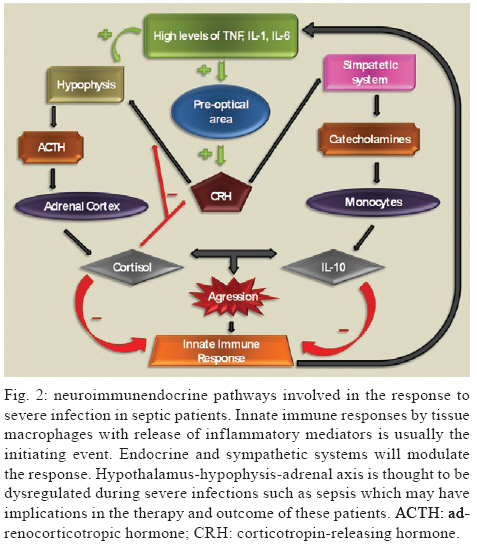
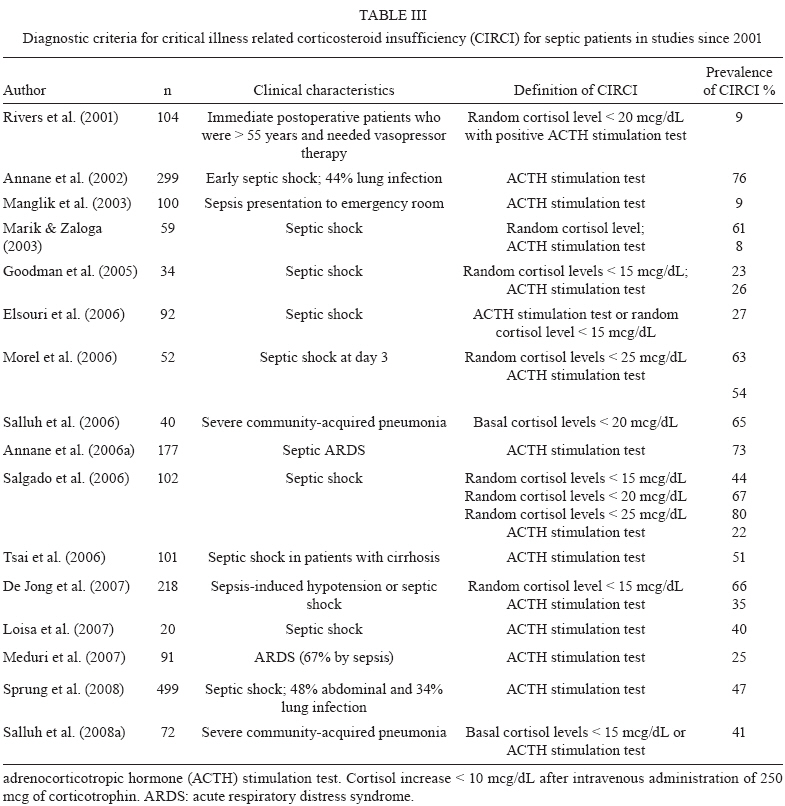
2009 Code Number: oc09126 ABSTRACT Corticosteroids are widely used to treat a diversity of pathological conditions including allergic, autoimmune and some infectious diseases. These drugs have complex mechanisms of action involving both genomic and non-genomic mechanisms and interfere with different signal transduction pathways in the cell. The use of corticosteroids to treat critically ill patients with acute respiratory distress syndrome and severe infections, such as sepsis and pneumonia, is still a matter of intense debate in the scientific and medical community with evidence both for and against its use in these patients. Here, we review the basic molecular mechanisms important for corticosteroid action as well as current evidence for their use, or not, in septic patients. We also present an analysis of the reasons why this is still such a controversial point in the literature. Key words: sepsis - septic shock - glucocorticoids - therapeutics - bacterial infections Glucocorticoids are used in the treatment of a wide range of inflammatory, allergic, autoimmune and infectious conditions. They have been applied successfully in the clinical setting for more than half a century. Despite their unquestionable utility in the treatment of several diseases, glucocorticoids have serious unwanted effects and their use is still a matter of intense debate in several pathological conditions. The use of glucocorticoids for the treatment of sepsis is probably one of the most striking examples of this contentiousness in the medical community. Several questions remain regarding the use of steroids for the treatment of sepsis. Should they be used at all? What dose of corticosteroid is appropriate? When should treatment begin? How should the steroids be used? Which patients will benefit from the treatment? What are the important molecular mechanisms involved? These and many other questions remain unanswered in the debate about the use of corticosteroids in septic patients. In the present review, we will summarise the current knowledge of the molecular mechanisms and clinical aspects of corticosteroid use in sepsis. In a recent meta-analysis, Annane et al. (2004) found that treatment with full doses of corticosteroids did not significantly affect mortality, but the use of long courses of low dose corticosteroids decreased mortality at 28 days. However, a subsequent multicenter, randomised, double-blind placebo-controlled trial could not confirm this finding and showed that the corticosteroid hydrocortisone did not improve survival or cause the reversal of shock in patients with septic shock (Sprung et al. 2008). Our aim in this review is not to answer the questions stated above, but rather to make them clear in order to reinforce the need to answer these questions and to spur the undertaking of additional studies that can point us in the right direction. History The era of glucocorticoids began with Thomas Addison, who graduated as a medical doctor from the University of Edinburgh, in 1815. Addison became interested in skin diseases and eventually described the alterations in skin pigmentation that are characteristic of Addison's disease. In conjunction with Dr. Richard Bright, Addison studied patients dying with damage to their adrenal glands and reported his observations in a lecture titled "Anemia - disease of the supra renal capsules" delivered in 1849 (Zimmerman 2007). Later on, experiments in the early 1900s demonstrated the key role of the adrenal glands in maintaining normal hemodynamics and selected cases of shock were found to be associated with haemorrhage of the adrenal gland (Brooks & Blalock 1934). In 1930, different investigators reported the use of adrenal cortex extracts to relieve adrenal insufficiency symptoms in both adrenalectomised animals and patients with Addison's disease (Hartman & Brownell 1933). Three years later and again in Science, Swingle et al. reported that adrenal cortex extracts were effective in treating shock, restoring circulating blood volume and kidney functions, decreasing haemoconcentration and increasing blood pressure. Also during the 1930s, numerous adrenal steroid compounds were isolated and chemically identified, leading to the identification of cortisone as a novel compound in 1936 (Mason et al. 1936). By 1948, sufficient quantities of cortisone had been isolated and used in small clinical investigations (Sarrett 1948). Several years later, additional studies demonstrated that corticosteroid replacement improved survival in adrenalectomised dogs challenged with lethal doses of bacteria and treated with standard antibiotics (Hinshaw et al. 1979, 1980). The first clinical evidence for the use of therapeutic corticosteroids in severe generalised infections appeared in 1951 (Hahn et al. 1951). A study that enrolled paediatric and adult subjects with severe infection was published in 1954 and the authors noted that "There is no question that the administration of ACTH or cortisone in sufficient amounts to patients with severe infections will result in rapid and striking clinical improvement." (Jahn et al. 1954). After those initial observations, multiple small clinical investigations were conducted, but it was only in 1963 that Bennett et al. published the first prospective, randomised placebo-controlled trial of hydrocortisone in sepsis (Bennett et al. 1962). Based primarily on results of impressive animal experiments (described previously) and initial clinical studies, corticosteroids were routinely used for adjunctive treatment of sepsis in 1960s. However, in the past 48 years we have witnessed a growing debate about if and how corticosteroids are useful in sepsis and waves of pro and con evidence have turned the issue of corticosteroid use for treating septic patients into an almost religious dispute. We will next summarise the basic molecular mechanisms of action and current evidence for the use of corticosteroids in patients with severe infections and sepsis. Molecular mechanisms of corticosteroid action Different studies have shown that corticosteroids activities can be divided into the classical genomic effects, mediated by the cytosolic glucocorticoid receptor (GR) and the more recently identified non-genomic effects (Pratt & Dittmar 1998, Almawi & Melemedjian 2002, Buttgereit & Scheffold 2002, Adcock & Lane 2003). These non-genomic corticosteroid activities can be further divided in three modes of action: GR-mediated non-genomic effects, non-specific non-genomic effects and effects that are considered to be mediated by membrane-bound GRs (mGCR) (Table I) (Buttgereit & Scheffold 2002, Spies et al. 2006). GR mediates the classical genomic actions of corticosteroids The GR is classically described as a 94-kD protein member of the steroid-hormone-receptor family and is expressed ubiquitously, although several isoforms generated by differential splicing or translation have been identified (Pratt & Dittmar 1998, Almawi & Melemedjian 2002, Wikstrom 2003). The GR consists of three domains with different functions: an N-terminal domain containing transactivation functions, a DNA-binding domain with a zinc-finger motif that is common to DNA interaction proteins and a ligand-binding domain consisting of 12 alpha-helices, which is involved in the formation of the hydrophobic ligand-binding pocket (Wikstrom 2003). Corticosteroids can easily pass through the plasma membrane due to their highly lipophilic characteristics. In its inactive form, the receptor is located in the cytoplasm of cells associated with molecular chaperones and immunophilins and is able to bind different corticosteroids with high affinity. After the binding of ligand to the GR, the receptor dissociates from this complex and is transported into the nucleus to modulate the expression of specific genes either positively (transactivation) or negatively (transrepression) (Pratt 1998, Almawi & Melemedjian 2002, Wikstrom 2003). Molecular chaperones shape responses to corticosteroids Chaperones facilitate the initial folding, maturation and, in some cases, regulate function of various proteins referred to as clients. Two main chaperone systems, the Hsp70 and Hsp90, influence GR assembly and activity. Hsp70 and Hsp90 are abundant, mainly cytosolic proteins. Hsp70 binds short, hydrophobic peptides and assists in the folding of nascent chains, while Hsp90 binds prefolded or even completely folded proteins and helps them to achieve or to maintain their active states (Grad & Picard 2007). Efficient maturation of GR to a conformation capable of high-affinity hormone binding requires Hsp70, Hsp40, Hsp90, Hop and p23 (Pratt & Dittmar 1998). The first molecular chaperone system that recognises de novo synthesised GR is the Hsp70 complex (Smith & Toft 1993). Hsp70 binds nascent proteins by recognizing hydrophobic segments of unfolded polypeptides in an ATP-regulated process. Hsp40 has also been shown to be a crucial component of the GR "foldosome" complex in reticulocyte extracts (Dittmar et al. 1998). It accelerates ATP hydrolysis by Hsp70, which results in tight binding of the substrate (GR) to Hsp70, which facilitates the binding of the next component of the complex, the tetratricopeptide repeat (TPR)-containing the cochaperone, Hop. Hop contains three TPR domains, two of which allow the simultaneous interaction with Hsp70 and Hsp90 and therefore allow a transfer of the substrate from the Hsp70 to the Hsp90 system (Grad & Picard 2007). Hsp90 regulates the final maturation of GR, facilitating a hormone-activatable state, which is dependent on ATP binding to Hsp90 (Grenert et al. 1999). GR has a 100-fold lower affinity for corticoids without Hsp90, as was shown in cell-free steroid binding assays (Nemoto et al. 1990). The affinity of Hsp90 for Hop is lower in the presence of ATP. Hop can then exit the complex making the TPR binding domain of Hsp90 available for the TPR-containing immunophilins, FKB52, FKBP51 and cyclophilin 40 and other cochaperones modulating GR-Hsp90 activity. Once bound to corticoids, GR has to move to the nucleus to function as a transcription factor. This step is also controlled by the Hsp90 machinery, specifically by the recruitment of immunophilin FKBP52 to the GR-Hsp90 complex. However, the details of the nuclear transport of GR remain controversial and incompletely understood (Grad & Picard 2007) (Fig. 1). Transactivation Positive regulation of gene expression by the GR has been shown to be mainly mediated by the direct binding of receptor homodimers to specific sequences [glucocorticoid responsive elements (GREs)] in the promoter regions of target genes. Three different types of GREs, simple, composite or tethering have been described (Lefstin & Yamamoto 1998), suggesting different molecular mechanisms for GR interaction with GREs. Activation of gene transcription by the GR via simple and composite GREs is dependent on the direct binding of the activated GR homodimer to DNA, although dimerization may not be required in composite GREs. On the contrary, GR binds to other DNA-bound transcription factors such as AP1, Stat3 and NFkB to increase transcription activity at tethering GREs. Regulation of gene transcription by GR requires the recruitment of coregulators. Among these coregulators, the p160 steroid receptor coactivator (Src) gene family contains three homologous members (Src-1, Src-2 and Src-3) that are crucial in facilitating chromatin remodelling, assembly of general transcription factors and transcription of target genes by the recruitment of histone acetyltransferases and methyltransferases to specific enhancer/promoter regions (Xu & Li 2003, Chinenov & Rogatsky 2007). Transrepression Different mechanisms account for negative regulation by the GR (Fig. 1). One of the best described is regulation via negative GREs that differ in structure and function from positive GREs (Dostert & Heinzel 2004). Such elements can be found at different positions in promoter regions and interfere either with the binding sites of other transcription factors or with the binding site of the basal transcription initiation complex (Schacke et al. 2007). However, only a few genes are known to be regulated via these negative GREs and the contribution of this kind of negative regulation to the overall activities of corticosteroids is probably of minor importance. The major mechanism of negative regulation by the GR is the inhibition of the activity of other transcription factors by being tethered to these factors. Interestingly, this mechanism is thought to occur through GR monomers (Heck et al. 1994, Reichardt et al. 1998). The transcription factors to which the GR binds include activator protein 1 (AP-1), nuclear factor-kB (NF-kB) and interferon regulatory factor 3 (IRF3) (Ogawa et al. 2005, Reily et al. 2006). Adding complexity to the system, Bladh et al. (2005) demonstrated that different domains of the GR seem to be responsible for either NF-kB or AP-1 interactions. A GR (R488Q) mutant unable to repress NF-kB activity retained the ability to repress AP-1 activity while transactivation activities were unaffected. Because all of these transcription factors regulate the expression of pro-inflammatory genes, their negative regulation by the GR has been considered the hallmark for the anti-inflammatory and immune suppressive action of corticosteroids. Other mechanisms identified that may help explain the transrepressive action of the GR is its binding to Jun N-terminal kinase (JNK) leading to suppression of JNK activity and, subsequently, to the inhibition of AP-1 (Caelles et al. 1997, Bruna et al. 2003) Cofactors It has been known for quite some time that GR does not function alone as a transcription factor. To achieve highly coordinated regulation of gene transcription, cofactors that modify and remodel chromatin structures are needed. Cofactors can be categorised as positive regulators of transcription (coactivators) or negative regulators of transcription (corepressors) depending on their function (Rogatsky & Ivashkiv 2006, Feige & Auwerx 2007). These cofactors do not bind directly to DNA, but are recruited through protein-protein interactions between transcription activation domains of the GR and regulatory sequences where they exert enzymatic activities such as histone acetylases (HATs) or deacetylases. Among cofactors that function as coactivators are CBP/p300, HATs and p160 proteins, including TIF2/GRIP1/NCoA3, pCIP/RAC3/ACTR/AIB1/NCoA3 and Src1/NCoA1 (Xu & O´Malley 2002). On the other hand, TIF2/GRIP1 is also able to function as a corepressor of the GR at the AP-1 and NF-kB tethering sites (Rogatsky et al. 2001). Despite the fact that the repression of many pro-inflammatory molecules seems to be a major part of the anti-inflammatory effects of corticosteroids, detailed knowledge about the molecular mechanisms involved is still scarce. For instance, additional corepressors, such as NCoR and SMRT, have also been described as being recruited to GR, although their role in the negative regulation of pro-inflammatory genes is not well recognised (Schacke et al. 2007) Rapid onset effects of corticosteroids are mediated by non-genomic mechanisms Mounting evidence suggests that corticosteroid effects cannot be entirely accounted for by the genomic mechanism of action explained above. Some effects are produced in a very short time, vanish rapidly and do not involve protein synthesis. Interestingly, some evidence suggests that these mechanisms are more ancient than the genomic ones, despite the fact that the latter were discovered earlier (Dallman 2005). There are three proposed non-genomic based mechanisms to explain the rapid anti-inflammatory and immunosuppressive effects of corticosteroids that are not compatible with the genomic mechanism of action (Croxtall et al. 2000, Buttgereit & Scheffold 2002, Hafezi-Moghadam et al. 2002): (i) non-specific interactions of corticosteroids with cellular membranes; (ii) non-genomic effects which are mediated by the cytosolic GR and (iii) specific interactions with a mGCR. Non-specific interactions of corticosteroids with cellular membranes - Plasma and mitochondrial membranes are targets for corticosteroids. At high concentrations corticosteroids intercalate into membranes and change their physicochemical properties as well as the activities of membrane-associated proteins (Buttgereit & Scheffold 2002, Buttgereit et al. 2004). As a consequence, calcium and sodium cycling across plasma membranes of immune cells is reduced and might contribute to rapid immunosuppression and decreased inflammation (Buttgereit & Scheffold 2002). In addition, proton leakage in the mitochondrial membrane was shown to be increased by glucocorticoids, resulting in impaired ATP production. This effect may contribute to clinically relevant outcomes in immune cells affecting cytokine synthesis, migration and phagocytosis, in conditions such as sepsis (Stahn et al. 2007) and may explain why high doses of corticoids can be harmful to septic patients. Non-genomic effects mediated by the cytosolic GR - As describe above, the unbound GR is located in the cytoplasm as a multi-protein complex consisting of heat-shock proteins and several other cochaperones. After corticosteroid binding, the GR dissociates from some of the proteins in this complex resulting in GR nuclear translocation. Some proteins on this complex, such as Src and MAPK, once released, can exert their signalling effects and therefore mediate the rapid non-genomic actions of the corticosteroid. For instance, the release of arachidonic acid from membrane-associated phospholipids is controlled by different proteins, such as MAPK and Phospholipase A2 and can be inhibited by corticosteroids by a GR-dependent, but transcription-independent mechanism (i.e., not blocked by transcription inhibitors such as actinomycin D) (Croxtall et al. 2000) in addition to the classical genomic-dependent effect. Specific interactions with a mGCR - The existence of a membrane bound corticosteroid receptor was first shown in lymphoma and leukemia cells (Gametchu et al. 1999). This receptor was later identified on human peripheral blood mononuclear cells through high-sensitivity immunofluorescent staining. Overexpression of GR did not cause increased mGCR expression on the cell surface. This indicates that mGCR is not just an unchanged GR that has been transported to the cell membrane. One possibility is that the mGCR is a variant of GR produced by differential splicing or promoter switching or by post-translational editing (Bartholome et al. 2004), but its origin still remains unexplained. It has been reported that stimulation with lipopolysaccharide (LPS) increases the percentage of mGCR-positive monocytes, suggesting that immunostimulation is responsible for up-regulation and transcellular transport of mGCR (Bartholome et al. 2004, Stahn et al. 2007). In addition, patients with rheumatoid arthritis, ankylosing spondylitis and systemic lupus erythematosus were shown to have high/increased numbers of mGCR-positive monocytes (Buttgereit et al. 2005, Spies et al. 2006, Tryc et al. 2006). The correlation between immune stimulation and increased expression of mGCR may have a clear impact on the importance of non-genomic effects for the corticosteroid actions in patients at an early phase of sepsis. This possibility, however, remains to be explored in future studies. More recent work has shed light on the molecular mechanism of the non-genomic mGCR-mediated immunosuppressive effects of corticosteroids in T cells. p56lck (Lck) and p59fyn (Fyn) kinases, members of the Src family of tyrosine kinases, were identified as cellular targets for non-genomic corticosteroid activities (Lowenberg et al. 2005). Lck and Fyn are expressed in T cells and they are involved in T cell receptor (TCR)-mediated signal transduction. Their association with the TCR complex is essential for efficient TCR signalling (Palacios & Weiss 2004). Lowenberg et al. (2005) have recently demonstrated that corticosteroid treatment rapidly inhibits Lck and Fyn activities in vitro and in vivo, via a mGCR-dependent pathway. These observations add complexity to the molecular mechanism of immune suppression by corticosteroids and indicate that a specific non-genomic mechanism may be of central relevance to corticosteroid-impaired TCR signalling. Corticosteroid effects on innate immune responses Innate immune responses play a central role in the pathophysiology of sepsis and are markedly influenced by corticosteroids and other endocrine and neural systems (Fig. 2). Mammalian cells sense the presence of pathogens through a family of transmembrane and cytoplasmic receptors, which detect conserved microbial components [lypopolysaccharide (LPS), peptidoglycans, single-stranded and double-stranded DNA and RNA and others) collectively referred to as pathogen-associated molecular patterns (PAMPs). This family of receptors is known as a group of pattern recognition receptors. Toll-like receptors (TLR) are one of the best characterised members of this family. To date, there are 10 members of the TLR subfamily that have been identified in humans. TLRs recognise a wide variety of PAMPs and upon activation, initiate a cascade of signalling events through several adaptor proteins and protein kinases that converge on the transcriptional regulators, NFkB, AP1 and IRFs, which, in turn, induce transcription of diverse cytokines and chemokines (Chinenov & Rogatsky 2007). The TLR signalling network is a target for GR-mediated actions and different mechanisms are involved in this effect. One of the most characterised mechanisms is the induction of endogenous inhibitors of the TLR signalling pathway. The induction of IkB by GR is a classic example of this mechanism (Scheinman et al. 1995), but other relevant targets were later revealed. JNK and p38 stress kinase pathways are activated by all known TLRs. Negative regulation of these pathways involves dephosphorylation of JNK, p38 and their upstream kinases, MEK3,4,6, by MAPK phosphatases (Abraham & Clark 2006). Interestingly, glucocorticoids inhibit JNK and p38 without altering their levels, suggesting that GR does not repress transcription of those kinases (Caelles et al. 1997, Hirasawa et al. 1998). One possible explanation is that GR induces transcription of an inhibitory phosphatase, MKP1, as already described in mast cells and endothelial cells (Kassel et al. 2001, Furst et al. 2007). In fact, in MKP1-/- mouse macrophages, glucocorticoid treatment failed to inhibit LPS-induced JNK and p38 activation and the production of inflammatory mediators (Abraham & Clark 2006). In addition, the MKP-1-/- mouse is extremely sensitive to LPS challenge and partially insensitive to the anti-inflammatory effects of corticosteroids (Chi et al. 2006, Wang et al. 2008). The precise mode of GR action on MKP1 gene transcription and to what extend MKP1 induction contributes to the anti-inflammatory actions of corticosteroids in in vivo models and in the clinical setting is not yet determined. Cytokines signal through the JAK/STAT pathways, which are sensitive to inhibition by the suppressors of cytokine signalling (SOCS) (Yoshida et al. 2004). In addition, SOCS1 also directly inhibit TLR 2 and 4 signalling by inducing rapid degradation of their TIR domain-containing adaptor TIRAP (Mansell et al. 2006). Corticosteroids induce SOCS1 mRNA in hematopoietic cell lines and in acute lymphoblastic leukemia patients. The precise mechanism of this up-regulation is presently unknown, but several potential GREs are scattered throughout the SOCS1 regulatory region and may play a role in corticosteroid-induced SOCS1 expression (Chinenov & Rogatsky 2007). Yet another potential mechanism for corticosteroid-induced immune modulatory effects involves glucocorticoid-inducible leucine zipper (GILZ), a small protein with a wide spectrum of anti-inflammatory activities, including the inhibition of LPS-induced expression of TLR2 in macrophages and of TLR 2 and 3-mediated NFkB activation in airway epithelial cells (D'Adamio et al. 1997, Ayroldi et al. 2001, Eddleston et al. 2007). GILZ also seems to be involved in enhancement of spontaneous thymocyte apoptosis, likely through down-regulation of the anti-apoptotic factor, Bcl-X (Delfino et al. 2004). GILZ is markedly induced by corticosteroids in a wide range of cell types by a mechanism that requires GR dimerization and DNA binding (Tonko et al. 2001, Berrebi et al. 2003, Rogatsky et al. 2003), suggesting that GILZ is a direct transcriptional target for the receptor. Indeed, several GREs were identified in the GILZ 5'-regulatory region (Wang et al. 2004, Chen et al. 2006). GILZ knockdown by siRNA prevents glucocorticoid inhibition of IL-1b-induced chemokine production in airway epithelial cells, suggesting that at least some of GR's anti-inflammatory actions require GILZ (Eddleston et al. 2007). Importantly, GILZ was also reported to physically interact with several effectors of TLR signalling, most notably Fos, Jun and p65/RelA (Ayroldi et al. 2001, Mittelstadt & Ashwell 2001). Experiments with GILZ knockout mice, presently unavailable, might provide some additional information on the role of this protein in the regulation of inflammation by GR. As described throughout this section, corticosteroids have many different mechanisms by which they interfere with innate immune responses. The intricate nature of corticosteroid effects can be easily illustrated by the apparent paradox involving TLR2 overexpression. Hyperactivation of TLRs, including TLR2, is thought to play an important role in excessive proinflammatory cytokine production and the pathogenesis of sepsis. Expression of TLR2 in response to TLR agonists is activated by NFkB (Liu et al. 2000, 2001) and could, therefore, be expected to be corticosteroid-sensitive. Surprisingly, several reports show corticosteroid induction of TLR4 and TLR2 mRNAs in multiple cell types (Shuto et al. 2002, Homma et al. 2004, Rozkova et al. 2006) through an incompletely known mechanism (Chinenov & Rogatsky 2007). While this seems like a paradoxical effect in light of corticosteroid anti-inflammatory effects, recent discovery of TLR2 and TLR4 expression in the adrenals (Bornstein et al. 2004a, b) may offer a potential explanation, since LPS and lipoteichoic acid directly stimulate cortisol release in adrenal cells (Vakharia & Hinson 2005). Furthermore, cortisol release upon TLR stimulation is impaired in TLR2 and TLR4-deficient mice (Zacharowski et al. 2006). Conceivably, a positive feedback loop is activated upon exposure to TLR ligands with the net output being the increased release of corticosteroids into the bloodstream, which would trigger classic mechanisms of inflammation reversal. As a net clinically observed effect, steroid infusion is able to modulate the inflammatory response in septic shock, as demonstrated by decreased levels of IL-6, IL-8, E-selectin and monocyte HLA-DR levels in a crossover study (Keh et al. 2003). Hydrocortisone was given early or late in different groups of septic shock patients and the function of monocytes and these cytokines were significantly influenced by steroid administration. Corticosteroid effects on vascular reactivity The complexity of cellular and molecular events involved in the vascular hyporeactivity that occurs in septic shock is far from being completely clarified. A variety of factors seems to be involved, including cytokines, platelet activating factor (Etienne et al. 1986), prostacyclin (Halushka et al. 1985), complement-derived C5a anaphylatoxin (Smedegård et al. 1989) and NO (Thiemermann & Vane 1990, Kosaka et al. 1992), all contributing to the hypotensive state and catecholamine hyporeactivity observed in septic shock. ATP-sensitive potassium (KATP) channels have been implicated in both hypotension and vascular hyporeactivity in septic conditions (Standen et al. 1989). In this context, Landry and Oliver (1992) have shown the involvement of KATP channels in the hypotension that occurs in the early phase of LPS septic shock in dogs, while Sorrentino et al. (1999) have demonstrated in the rat that an increase in KATP channel activity is implicated in the vascular hyporeactivity to contracting agents observed in the delayed phase of LPS-induced shock. The involvement of KATP channels in septic shock was later confirmed by Czaika et al. (2000), who demonstrated an up-regulation of u-K(ATP)-1 protein expression in this pathological condition. Also, another type of potassium channel, the calcium-activated potassium channel, has been shown to have a role in in vitro and in vivo models of hyporesponsiveness to catecholamines (Chen et al. 1999, Terluk et al. 2000). In fact, GR agonists inhibit the expression of calcium-dependent potassium channel protein in primary vascular smooth muscle cell cultures (Brem et al. 1999), suggesting that corticosteroids could be inhibiting potassium channel protein expression and/or the synthesis of a mediator that would regulate the KATP channel expression. More recently, dexamethasone was shown to improve the vascular hyporeactivity to catecholamines in LPS-treated rats due to an inhibitory effect on KATP channel activity (d'Emmanuele di Villa Bianca et al. 2003). This result fits with the clinical observation that hydrocortisone treatment in septic shock patients improves the response to adrenergic agonist infusion (Bellissant & Annane 2000) and that facilitates shock reversal (Annane et al. 2004). Clinical trials evaluating corticosteroid administration in sepsis and acute respiratory distress syndrome (ARDS) Corticosteroids have been given to patients with severe infections for more than 90 years, as described by Friderichsen (1918) in hemorrhagic necrosis of adrenal glands in children with purpura fulminans (Waterhouse-Friderichsen syndrome). An association with meningococcemia was done by the same author after almost 40 years. Since then, corticosteroids have been used in the treatment of a myriad of severe infections: Haemophilus influenzae and Streptococcus pneumoniae meningitis, herpes zoster, tuberculosis (meningitis, ophthalmic or pericardial commitment), typhoid fever, infectious mononucleosis, Pneumocystis pneumonia, tetanus, hepatitis B, pertussis and acute bronchiolitis, just to name a few (McGee & Hirschmann 2008). Many patients with severe infections also have clinical shock and the reduction in arterial pressure and tissue perfusion is associated with microbial load and severity of illness. Severe bacterial infections cause systemic inflammatory response defined as sepsis in earlier consensus conference (Bone 1992). Because of our understanding that sepsis is an exaggerated host response to infections, steroids were quickly proposed for its treatment. Table II summarises the most important results and notes about clinical trials of steroid therapy for severe sepsis and septic shock. Steroids became a standard treatment for severe infections between the 1960s and 70s. Some small trials showed consistent findings that steroid treatment caused faster reversal of shock and lower mortality rates. Schumer (1976) published a trial with more than 300 patients with an almost 60% reduction in mortality rate among those treated with dexamethasone or methylprednisolone. However, like this study, most of the studies only included a small population, were poorly controlled or were retrospective trials. In the early 80s, randomised clinical trials analysed the administration of high doses of methylprednisolone (30 mg/kg/day). It was preconized that corticosteroid therapy should begin early, usually on the first day of septic shock and it was rarely sustained beyond this time. Three studies were representative of this strategy. Sprung et al. (1984) enrolled 59 patients with no more than 24h of septic shock and treated them with methylprednisolone, dexamethasone or placebo. Although the reversal of shock in 24 h was more evident in both steroid groups, mortality was not significantly different. Bone et al. (1987b) used methylprednisolone in 382 patients with severe sepsis (most of them presented septic shock) and found that there was no benefit to the steroid treatment. In fact, more harm than good was observed in the renal insufficiency subgroup. Finally, Luce et al. (1988) used methylprednisolone or placebo in acute lung injury patients; the majority of patients had concomitant septic shock. The main goal was the prevention of ARDS, but there was no benefit in the degree of parenchymal lung injury or mortality. By the negative results of these major trials, corticosteroid treatment in sepsis was restricted to rare selected cases for more than a decade. Criticism was attributed to these studies by the fact that adrenal dysfunction could last more than 24-48 h, as originally described and large doses were administered, which could have contributed to the higher incidence of hospital-acquired infections as a consequence of profound immunosuppression. After almost 10 years, new evidence for the beneficial effects of steroid treatment was shown in small randomised trials. Most authors aimed to analyse the effects of low doses of hydrocortisone on the reversal of septic shock. Corticosteroids were also used for a prolonged time (usually 7 days) and tapered over several days. Bollaert et al. (1998) studied 41 patients with late refractory septic shock (more than 48 h of evolution) and randomised them to receive 100 mg of hydrocortisone or placebo for five days. The steroid group showed a significant improvement in shock reversal, although they showed no difference in regard to the response to corticotropin test. There was also a trend toward reduced 28-day mortality. Briegel et al. (1999) enrolled 40 patients within 24-28 h of septic shock to either receive or not receive an intravenous infusion of hydrocortisone. The group receiving the steroid infusion had reduced time to the cessation of vasopressor therapy (median 2 vs. 7 days, p < 0.01). In this trial steroid therapy was rapidly tapered after catecholamine withdrawal. Two other trials found similar results in reversal of shock and a trend toward reduced early mortality in patients with steroids (Chawla et al. 2000, Yildiz et al. 2002). These small randomised trials brought hope again to the usefulness of corticosteroids in critically ill septic patients and multicenter clinical trials with larger population were then planned to confirm this newer successful evidence. Most of recent evidence about the effect of cortico-steroids on reversal of shock and mortality come from two randomised trials: the French trial and Corticosteroid Therapy of Septic Shock (CORTICUS). The former study enrolled 299 patients with early septic shock (usually in the first 8 h) (Annane et al. 2002). It was placebo-controlled, randomised, double-blinded study performed at 19 intensive care units in France. Intravenous hydrocortisone and enteral fludrocortisone were used for seven days and the main outcome was mortality by day 28 after inclusion. The authors classified patients according to response to adrenal stimulation test, which consisted of intravenous administration of 250 mcg of the adrenocorticotropic hormone (ACTH) analogue corticotropin and measurement of serum cortisol after 1 and 2-h intervals. A patient was considered a non-responder (adrenal dysfunction) when there were increments in cortisol levels below 10 mcg/dL (ACTH test). Adrenal insufficiency was present in 229 non-responders according to this screening test. In this subgroup, 28-day mortality had an absolute 10% reduction. Reversal of shock was also achieved in 57% vs. 40% in the placebo group, suggesting the previous hypothesis of steroid increase in adrenergic receptor responsiveness. In contrast to these positive effects, responders to the corticotropin test had no benefits in steroid replacement. When adding the French study to other studies that have been conducted since 1998, steroids became the standard of care in early septic shock, as recommended by the worldwide task force to reduce sepsis mortality, the Surviving Sepsis Campaign, in 2004 (Dellinger et al. 2004). Hydrocortisone was given to septic shock patients, usually in the first 24 h after presentation, in doses of 200 to 300 mg per day for 7-10 days; fludrocortisone was not routinely recommended. In order to confirm the benefits of corticosteroid, a second large trial (CORTICUS) was conducted in which researchers in 11 European countries designed a randomised, placebo-controlled trial with septic shock patients (Sprung et al. 2008). Again, the main goal was 28-day mortality in non-responders to the corticotropin test. Hydrocortisone was administered as a 50 mg dose every 6 h for 11 days and tapering was done by day 5-11, in order to avoid rebound hypotension. Enrolment included 499 patients, of whom 47% were non-responders to the corticotropin test. Mortality on day 28 or in the hospital was not different between the two groups and the corticosteroid group had a higher incidence of superinfections (infection superimposed upon previous ones) and higher rates of new septic shock episodes. Hyperglycemia was also more frequent in the corticosteroid group. One possible explanation for these contrasting observations is that the mode of hydrocortisone administration could have an impact on glycemic control, as shown with bolus injections and a higher fluctuation in blood glucose levels would impact the outcome (Loisa et al. 2007, Weber-Carstens et al. 2007). These two randomised double-blinded clinical trials had good methodological quality, but their results were opposite. In both studies, there were problems in study design, as for example, low recruitment during enrolment, possibly because of the strict deadline by which to give the first dose of hydrocortisone. When analyzing their sample sizes, CORTICUS had less patients than previously calculated for 80% statistical power and a 10% absolute decrease in mortality (n = 800). In the French study (Annane et al. 2002), sample size was calculated for 270 patients (they actually included 299) and non-responders to the corticotropin test reached a significant 20% relative reduction in mortality. Some other differences are noteworthy: in the French trial, patients received steroids in the first 8 h of septic shock presentation, while in CORTICUS there was a looser deadline of 72 h for inclusion. Maybe for this reason, reversal of shock occurred more frequently in the former study. The severity of illness was calculated with a prognostic score (Simplified Acute Physiology Score II - SAPS II), which predicts hospital mortality based on demographic and physiologic data collected in the first 24 h after patient admission to intensive care unit (ICU). The score was higher in the former study by Annane, with a great difference between SAPS II scores: 58 vs. 48 points in CORTICUS. This difference denotes the different severity of these two studies populations. More severely ill patients could possibly benefit more from immune therapy, as already shown in the case of other therapies (Bernard et al. 2001). Also, in the French study statistical analyses were done for 1-sided formulation, which means that the authors did not consider the adverse effects of steroids in the treatment of septic shock. Another possible reason was the difference in the site of infections: pneumonia was the major source of sepsis in the French study, while abdominal/peritoneal infections were the majority in CORTICUS. There is evidence supporting benefits to the use of corticosteroids in pneumonia and cortisol being a biomarker of prognosis in this infection (Confalonieri et al. 2005, Salluh et al. 2008a), while abdominal infections are usually dependent on maintenance of the macrophage population in the peritoneum, antibiotic penetration in necrosis and surgical intervention (Hausmann et al. 2000, Sotto et al. 2002). In addition to the differences in the sites of infection, appropriate antimicrobial therapy was more prevalent in the CORTICUS placebo group (78.8% vs. 72.8%), which can obviously lessen the rate of success in corticosteroid therapy. Patients received corticosteroids for longer periods in CORTICUS (11 days), even if they recovered from shock and hyperglycemia and new infections were far more frequent than in previous studies. Annane et al. (2004) performed a meta-analysis of steroid use in septic shock in 2004 and he identified that, even in older studies (high-dose and short duration), incidence of superinfections and serum glucose levels were not altered by intervention. CORTICUS was unique in showing that longer therapy is associated with adverse events, which could offset eventual benefits of the anti-inflammatory effects of corticosteroids. An international task force was created to develop a consensus statement for the diagnosis and management of corticosteroid insufficiency in critically ill patients and a meta-analysis demonstrated that the addition of CORTICUS data to the earlier studies confirmed a greater rate of shock reversal up to seven days of septic shock, although there was no effect on the 28-day mortality (Marik et al. 2008). Early reversal of shock was achieved in almost 65% of all patients in the low-dose steroid studies, while only 47.5% presented the same response in the placebo group (relative risk: 1.39, 95% confidence interval: 1.24-1.55). Mortality after one month was not lowered by corticosteroid administration (relative risk: 0.92, 95% confidence interval: 0.79-1.06). The task force recommended corticosteroid treatment in septic shock to patients with poor response to fluids and vasopressor, irrespectively of any diagnostic test of critical illness-related corticosteroid insufficiency (basal total or free serum cortisol or rising cortisol levels after synthetic ACTH administration). Several data are available from clinical and experimental studies that provide a sound physiologic rationale for the use of corticosteroids in ARDS (Calfee & Matthay 2007). However, translation of this scientific background into unequivocal clinical evidence is still a promise to be fulfilled (Steinberg et al. 2006). Evidence of excessive pro-inflammatory stimuli in the alveolar compartment coupled with biochemical evidence of fibroproliferative activity are available even in the earliest stages of ARDS (Chesnutt et al. 1997a, b, Rocco et al. 2003, Fernandes et al. 2005, Santos et al. 2006). The current interpretation of data from clinical trials of corticosteroids in ARDS is significantly limited by the heterogeneity of drug administration protocols regarding the dose, timing, treatment duration and eligibility criteria employed in the studies (Bernard et al. 1987, Bone et al. 1987a, Steinberg et al. 2006). Two recently published meta-analyses evaluated the role of corticosteroid treatment in ARDS and reached different conclusions (Meduri et al. 2008, Peter et al. 2008). Nevertheless, it seems reasonable to ascertain that the use of high doses of corticosteroids is associated with significantly increased morbidity (Bernard et al. 1987, Bone et al. 1987a). Moreover, when corticosteroids were used in moderate doses in early severe ARDS (Annane et al. 2006b, Meduri et al. 2007) or in unresolving ARDS (Meduri et al. 1998) they are associated with improved outcomes. The use of corticosteroids as a rescue therapy in patients with persistent ARDS lasting for more than 14 days was evaluated in a large multicenter trial and no significant clinical benefit was observed in this population, but rather it increased risk of hospital-acquired infections (Steinberg et al. 2006). Moreover, pulmonary or extra-pulmonary ARDS also seem to respond differently to corticosteroid treatment, at least in pre-clinical studies (Leite-Junior et al. 2008). In the view of these studies, corticosteroids cannot be widely recommended for critically ill patients with ARDS. In recent years, the use of corticosteroids has been suggested to be effective in selected patients with community acquired pneumonia (CAP) (Confalonieri et al. 2005, Annane & Meduri 2008, Sibila et al. 2008). Nevertheless, recent reviews concluded that available studies cannot support recommendation for corticosteroids as standard of care for patients severe CAP (Salluh et al. 2008b, Sibila et al. 2008). In a recent study, we have studied 191 patients with CAP and concluded that in patients with severe CAP, adjunctive therapy with corticosteroids may facilitate the weaning of vasopressors but has no influence on mortality, on the clinical course assessed by the SOFA score or on the C reactive protein course. However, if necessary, corticosteroid prescription seems to be safe in these patients (JI Salluh et al., unpublished observations). Critical illness-related corticosteroid insufficiency (CIRCI) Any critical illness is able to blunt adrenal synthesis of cortisol or its action (increased resistance). Low levels of cortisol were demonstrated in sepsis, pancreatitis, severe burns and liver failure (Murphy et al. 1993, Marik et al. 2005, Widmer et al. 2005, Annane et al. 2006a, Ho et al. 2006). Decreased production of cortisol or ACTH is common in severe sepsis or septic shock, particularly in patients with risk factors, such as female gender, positive blood cultures or Gram-negative infections (Annane et al. 2006a, Salgado et al. 2008). Based on experimental data, severe inflammation-associated corticosteroid resistance is thought to primarily exist due to decreased corticosteroid receptor nuclear translocation after endotoxin or proinflammatory cytokine challenges (Liu et al. 1993, Pariante et al. 1999). Some interesting evidence came from a clinical study that demonstrated the same phenomenon in patients with ARDS; patients had severe gas exchange compromise and low baseline cytoplasmic and serum cortisol levels, indicating impairment of corticosteroid action (Meduri et al. 2005). CIRCI is considered the most correct term for inadequate relative adrenal response to a severe critical disease, mainly sepsis (Marik et al. 2008) and was formerly known as relative adrenal insufficiency or failure. Diagnosis of CIRCI in critically ill patients is based on two methods: random total serum cortisol or change in cortisol levels after synthetic ACTH administration (Table III). High levels of random serum cortisol (collected anytime from the beginning of sepsis) have been recognised as a sign of severe infection for many years. In a cohort of 20 patients with shock due to infection, it was observed that non-survivors had significantly higher serum cortisol than survivors (Melby & Spink 1958). Marik and Zaloga (2003) determined that any critically ill septic patient with a random serum cortisol of 25 mcg/dL or less has high probability of adrenal insufficiency. Admitting severe infections in the emergency room can also reveal a subset of early CIRCI patients. Their presentation is somewhat different from other severe sepsis patients, with lower levels of glucose and sodium and baseline ACTH and cortisol levels (Manglik et al. 2003). The synthetic ACTH stimulation test is advocated for a more specific diagnosis of CIRCI. It can be done with a full dose (250 mcg) or a low dose (1 mcg). Annane et al. (2000) conducted this test in a large cohort of early septic shock patients and could identify a subset of higher death probability group with a poor response to synthetic ACTH and higher levels of basal serum cortisol. Better prognosis was shown in patients with basal cortisol less than 34 mcg/dL and delta cortisol after ACTH test higher than 9 mcg/dL (26% mortality). The other response patterns were associated with significantly higher mortality: basal cortisol higher than 34 mcg/dL and/or delta cortisol less than 9 mcg/dL patients had only 18-33% survival. Bollaert et al. (2003) also obtained a similar result with late (around 4 days) septic shock patients: basal cortisol > 20 mcg/dL and delta cortisol < 9 mcg/dL were independent predictors of mortality. CIRCI can also be a dynamic process. Adrenal exhaustion is thought to be a different form of CIRCI and septic shock patients can exhibit normal or high basal cortisol levels at hospital admission, but adrenal response can become severely altered after a few days. After retesting a cohort of septic shock patients with long vasopressor dependency, Guzman and Guzman (2007) confirmed that these patients presented very low levels of random serum cortisol levels after six days with normal or high cortisol levels at admission (from initial mean ~42 mcg/dL to repeated 10 mcg/dL levels). Hydrocortisone administration aided in vasopressor weaning in all of these patients (Guzman & Guzman 2007). Cortisol changes among septic shock patients is very dynamic and can persist for up to 28 days and there is wide difference in random cortisol and delta cortisol (synthetic ACTH test) between survivors and non-survivors (Goodman et al. 2005). These important data suggest that accompanying cortisol levels in vasopressor-dependent patients can be useful and sometimes dictate the need for a steroid course during septic patients' ICU stay. Conflicting information came onto the scene a few years ago, when it became clear that free levels of cortisol (the real bioactive form of the hormone) are highly influenced by serum protein levels, mainly albumin and cortisol-binding protein. Different levels of basal and ACTH-stimulated cortisol were seen in a cohort of 66 patients with septic shock, but free levels of the hormone were very similar when balanced with serum albumin (Hamrahian et al. 2004). Albumin levels of 2.5 g/dL were set as an important limit for analyzing serum cortisol levels. Confirming data came from another study, in which different levels of random cortisol levels were set with a 2.5 mg/dL serum albumin limit and it showed a good correlation with the synthetic ACTH stimulating test (Salgado et al. 2006). Recently, Christ-Crain et al. (2007) studied total and free serum cortisol levels in patients with severe community-acquired pneumonia and verified a similar performance in the prediction of the severity of disease, indicating that one can expect close significance concerning the severity of illness or mortality. Dosage of the free portion of serum cortisol can become very useful in critically ill patients, but it comes with some disadvantages, like augmenting costs and the need for setting specific levels for the diagnosis of CIRCI. Even when diagnosing CIRCI, corticosteroid treatment is not straightforward for improving septic shock management. Elsouri et al. (2006). analysed 92 patients with septic shock and CIRCI was present in almost half of them, based on a 250 mcg synthetic ACTH test. CIRCI patients needed rescue vasopressor therapy more frequently, but maximal doses of conventional catecholamines were similar to non-CIRCI patients. Besides, hydrocortisone therapy was given to only half CIRCI patients and did not affect mortality. Morel et al. (2006) made an overview of vasopressor-dependent septic patients and studied parameters associated with hemodynamic improvement after corticosteroid initiation (reduction of more than 50% in vasopressor dose up to 3 days of steroid treatment). They compared different diagnostic tests for CIRCI (random total cortisol or delta cortisol to 1 and 250 mcg synthetic ACTH test) and verified that hemodynamic improvement was not associated with diagnosis of CIRCI. Another retrospective study conversely stated that CIRCI diagnosed patients have a higher mortality if not treated with glucocorticoids, although this was a very small subgroup of patients (n = 7) (de Jong et al. 2007). Interestingly, possible CIRCI patients had similar survival when compared to non CIRCI patients who were also treated with corticosteroids. Based on these small retrospective data, making the diagnosis of CIRCI does not alter indications for steroid treatment in septic shock. When and how to use steroids in septic shock patients? Common sites of infection in critically ill patients are lungs, abdomen, catheter-related and the urinary tract. The severity of each of these sites is different and is thought to be higher in abdominal and pulmonary infections. Incidence of organ dysfunction and response to specific therapies are also different, as recently shown with recombinant activated protein C (Abraham et al. 2005, Sevransky et al. 2008). Another example is severe CAP, which was successfully treated with low doses of hydrocortisone in a small clinical trial, although certainty about its indication remained in low to moderate level of recommendation (Confalonieri et al. 2005, Salluh et al. 2008b). Conversely, severe abdominal infections present with a poor prognosis, since antibiotic tissue penetration and drainage of abscesses are often harder to guarantee. Urinary tract and venous catheters are also other common sites of infections in the critically ill and their treatment encompasses early antibiotic administration and device removal, with a generally rapid and good response. Therefore, different infections show diverse patterns of presentation and evolution, making it difficult to predict what is the best treatment for each situation. This reason may be explain why most anti-inflammatory therapies tried in septic patients such as corticosteroids, recombinant activated protein C, antithrombin, tifacogin, anti-TNF antibodies and polyclonal immunoglobulins just to name few, did not work as expected based on the initial pre-clinical studies. There are interesting data regarding the use of corticosteroids as an adjunctive treatment for different infections. One early example is the use of corticosteroids in hypoxemic Pneumocystis jiroveci pneumonia; a meta-analysis revealed that there is a reduction in need for mechanical ventilation and in 1 and 3-month mortality (Briel et al. 2005). There are only six clinical trials on this topic, with great heterogeneity and a diverse number of patients. Oxygenation entry criteria were also diverse (from PaO2 of 51-75 mmHg or oxygen saturation less than 85-90%). Intravenous methylprednisolone and oral prednisone were the tested routes of administration. The gap between the beginning of antibiotic treatment to first dose of corticosteroids ranged from 24 h to unlimited and there was no mention to tapering schedules. Nevertheless, steroids showed a clear benefit to these patients, lowering need for mechanical ventilation by more than 60% and mortality by 33%. This result means that in the treatment of at least nine P. jiroveci pneumonia patients, adjunctive steroid therapy will save one individual. Besides all of the critiques to the results of these trials, there is no disagreement in adding corticosteroids to hypoxemic P. jiroveci pneumonia patients. A recent systematic review concluded that, while corticosteroids cannot be recommended as a standard of care for patients with severe community-acquired pneumonia, studies that evaluated the use of moderate doses for 7-10 days demonstrated clinical benefits (Salluh et al. 2008b). It is also interesting to observe that significant derangements in the adrenal response have been described previously in patients with severe community-acquired pneumonia and is associated with worse outcomes (Salluh & Fuks 2006, Christ-Crain et al. 2007, Gotoh et al. 2008, Salluh et al. 2008a). Another excellent example is use of corticosteroids as adjunctive treatment in bacterial and tuberculous meningitis. Several clinical trials showed conflicting results and efficacious treatment depends on bacterial species, mainly with H. influenzae, Mycobacterium tuberculosis and S. pneumoniae (Lebel et al. 1988, de Gans & van de Beek 2002). Patients with meningococcal infections saw no benefit upon adding corticosteroids to routine treatment in these two large trials, although it is usually more severe in early clinical presentation than other forms of meningitis (van de Beek et al. 2007). Controversies continue in the literature, but corticosteroids are often given to comatose patients with meningitis, mainly with low glucose and high protein in cerebrospinal fluid, until bacterial identification is done. Severe cases of typhoid fever are classical indications for the administration of corticosteroids (Hoffman et al. 1984). Stratification of steroid need for these patients was done many years ago. Torpor and shock preclude severe presentations, which usually respond better to chloramphenicol and corticosteroid administration than to antibiotic alone, although mild and moderate cases do not benefit from the same strategy. On the other hand, corticosteroid treatment has no place in other several infections, such as cerebral malaria, acute bronchiolitis by respiratory syncytial virus or viral hepatitis. Treatment with corticosteroids can worsen their prognosis (Gregory et al. 1976, Warrell et al. 1982). When analyzing incidence and mortality from infectious diseases around the globe, we must take into account the very higher number of cases in undeveloped and in-developing countries (Cheng et al. 2008). Pneumonia and meningitis are the main causes of infectious disease-related morbidity and mortality in lower income locations, which are theoretically very different from higher income ones. Therefore, as higher quality clinical trials of steroid use in severe sepsis or septic shock came from Europe and the United States of America, data regarding this treatment can dramatically differ in the rest of the world. For confirming or denying this theory, clinical studies regarding corticosteroid administration in severe infections (and sepsis) must be applied to different parts of the globe. Besides looking at the role of steroids treatment in different sources of infection, some practical considerations can be made: (i) steroids must be used in septic shock patients and not in sepsis or severe sepsis without hypotension (Dellinger et al. 2004, Marik et al. 2008). There is no role for its anti-inflammatory effects and/or immunomodulation as shown in 1980s clinical trials. A "permissive" or catecholamine-adjunctive or role for steroids may present the main effect on septic shock patients; (ii) hydrocortisone is the most appropriate steroid drug to be used. It has equal glucocorticoid and more potent mineralocorticoid action than other usual steroids, such as methylprednisolone or dexamethasone, and may possibly lessen the need to use fludrocortisone. It should be given intravenously in 50-100 mg doses every 6-8 h; (iii) it seems reasonable to insist on giving fluids and low-dose vasopressors in the first 24 h of septic shock before initiating steroids, because many patients recover from this condition in a few hours or days (Rivers et al. 2001). Steroids may be useful in longer and/or high-dose vasopressor-dependent shock; (iv) any diagnostic test for CIRCI (total serum cortisol or ACTH test) may be useful for prognosis, but cannot be indicated for guiding steroid therapy, although very low levels of serum cortisol (below 10 mcg/dL) are indicative of adrenal failure; (v) serum levels of cortisol can be influenced by serum protein (particularly albumin) levels (Hamrahian et al. 2004); (vi) a positive response to steroid treatment generally corresponds to fast tapering of vasopressors (up to 3 days) and (vii) based on recent evidence, steroid therapy longer than one week can cause adverse effects, such as hyperglycemia and superinfections (Sprung et al. 2008). If there is a positive response, steroid can be tapered over few days; on the contrary, a negative response must not be indicated for longer administration and can be suspended up to 5-7 days. Concluding remarks Systemic corticosteroids have been used as adjunctive therapy for severe infections for more than 60 years; however, their prescription is still a source of intense controversy. Corticosteroid use in sepsis and acute lung injury was initially proposed to counteract the intense systemic and pulmonary inflammation. Different corticosteroid molecules (natural versus synthetic), dosage and therapy duration is a wide field of heterogeneity among many studies, opening possibilities for future researches. Perhaps one of the crucial aspects for testing pharmacologic interventions is patient selection. Acknowledging that point, severe sepsis encompasses a wide range of infections of distinct sources and etiological agents in heterogeneous groups of patients; therefore, it is difficult to generalise the results of any successful intervention as valid for all patients with severe infections. Besides, the inflammatory response to infection is not static, but is dynamic with marked changes in biomarkers and mediators over time. The assessment of the immune status (either hyper or hyporeactive), although crucial, remains unattainable with the currently available methods (Marshall et al. 2003, Bozza et al. 2005, 2007, Salluh et al. 2008b). More effective stratification of patients, along with more precise evaluation of the immune status of the patients, seem, to be critical to the success of corticosteroid therapy in the critically ill and should be pursued in future studies aimed to confirm or rebut the usefulness of corticosteroid in patients with severe infection. ACKNOWLEGDMENTS To Kelly Grace Magalhães, for her skilled help with preparations of figures, and to Adriana Vieira de Abreu Broxado, in organizing the references cited in this article. REFERENCES
Copyright 2009 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc09126f2.jpg] [oc09126f1.jpg] [oc09126t3.jpg] [oc09126t2.jpg] [oc09126t1.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}