 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 104, No. 8, 2009, pp. 1063-1071 ARTICLES Absence of Fas-L aggravates renal injury in acute Trypanosoma cruzi infection Gabriel Melo de OliveiraI, +; Masako Oya MasudaII; Nazaré N RochaIII; Nestor SchorIV; Cléber S HooperV; Tânia C de Araújo-JorgeI; Andréa Henriques-PonsI ILaboratório
de Biologia Celular, Instituto Oswaldo Cruz-Fiocruz, Av. Brasil 4365, 21045-900,
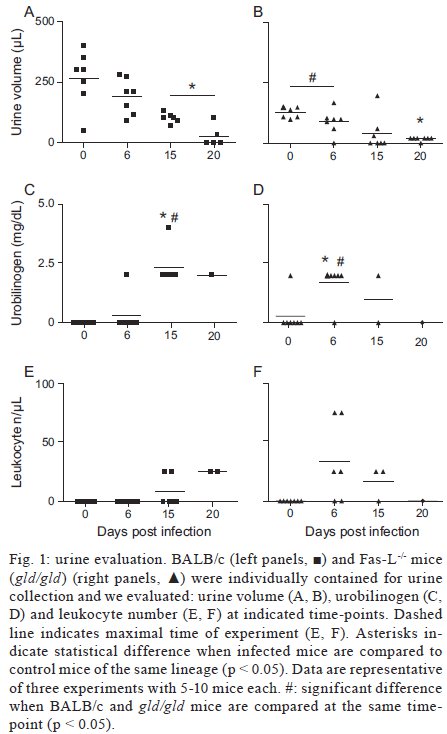
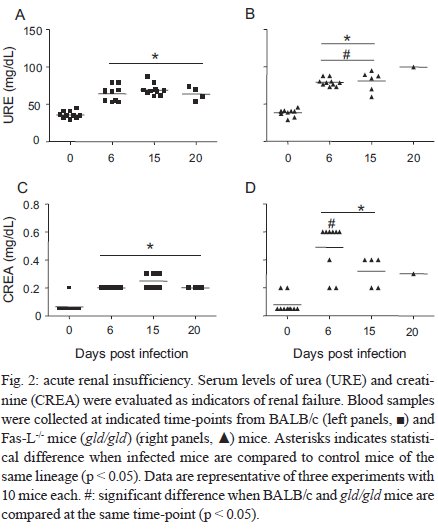
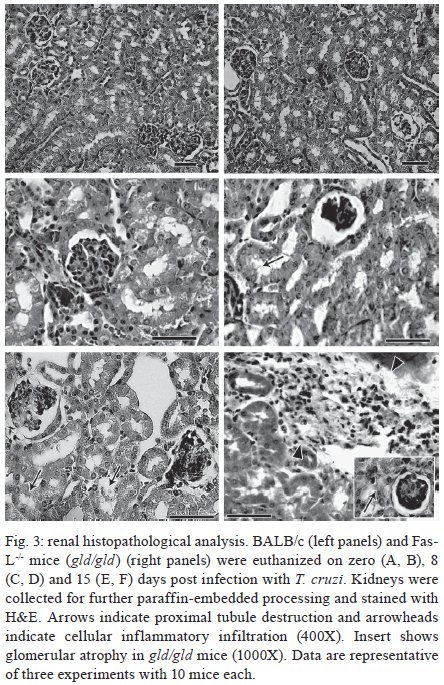
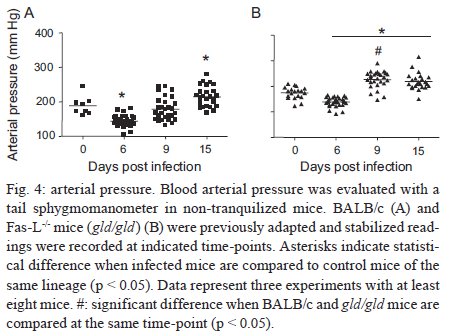
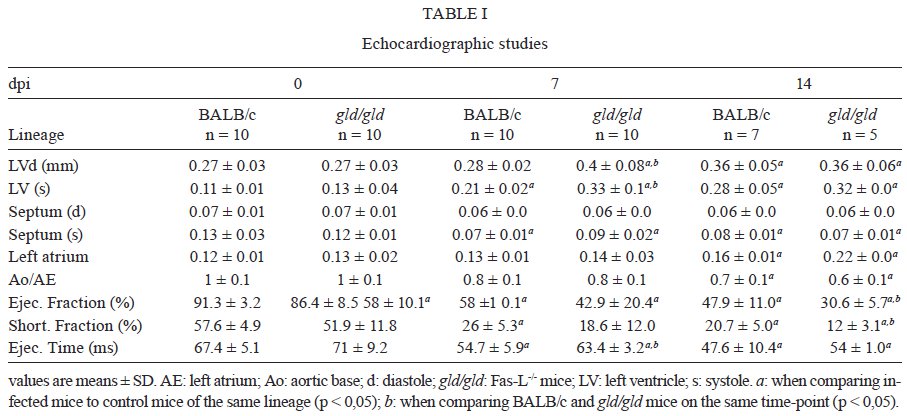
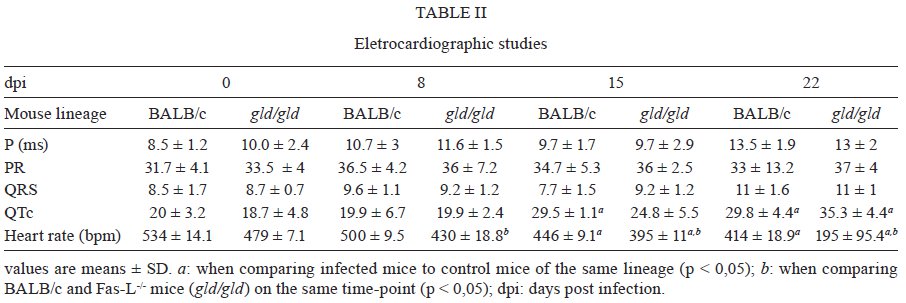
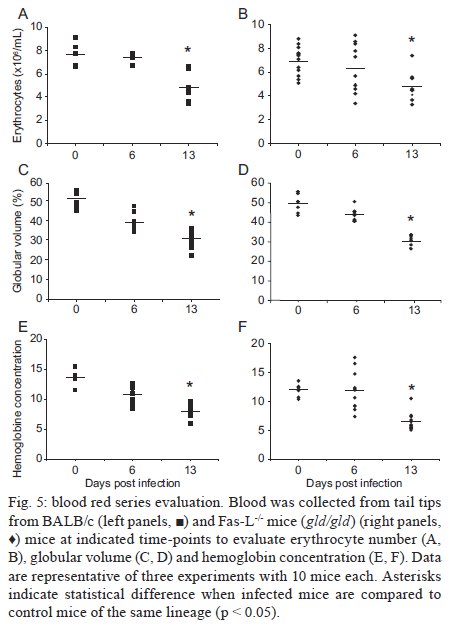
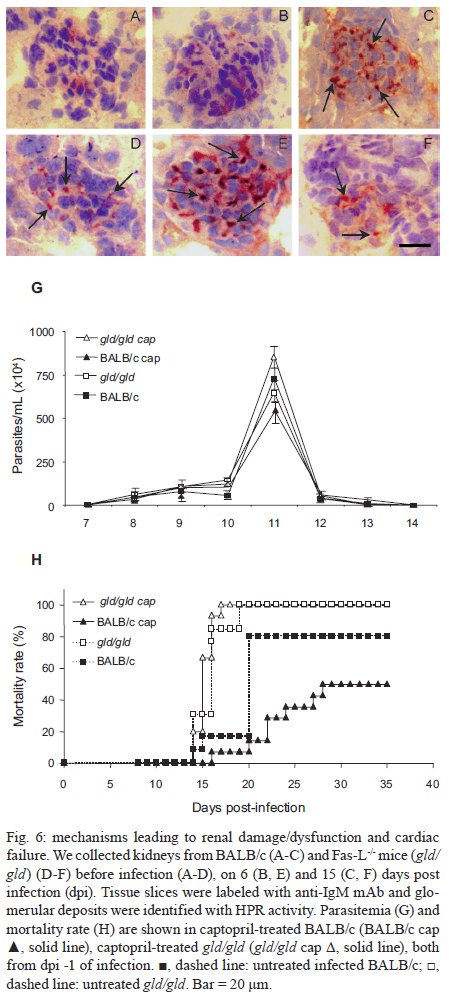
Rio de Janeiro, RJ, Brasil Received 11 March 2009 Code Number: oc09220 ABSTRACT Trypanosoma cruzi infection induces diverse alterations in immunocompetent cells and organs, myocarditis and congestive heart failure. However, the physiological network of disturbances imposed by the infection has not been addressed thoroughly. Regarding myocarditis induced by the infection, we observed in our previous work that Fas-L-/- mice (gld/gld) have very mild inflammatory infiltration when compared to BALB/c mice. However, all mice from both lineages die in the early acute phase. Therefore, in this work we studied the physiological connection relating arterial pressure, renal function/damage and cardiac insufficiency as causes of death. Our results show that a broader set of dysfunctions that could be classified as a cardio/anaemic/renal syndrome is more likely responsible for cardiac failure and death in both lineages. However, gld/gld mice had very early glomerular deposition of IgM and a more intense renal inflammatory response with reduced renal filtration, which is probably responsible for the premature death in the absence of significant myocarditis in gld/gld. Key words: Trypanosoma cruzi - Fas-L - myocarditis - acute kidney injury Chagas disease is caused by the protozoan parasite Trypanosoma cruzi, which has a widespread distribution in Latin America. It is estimated that 15-16 million people are infected on this continent and 75-90 million are exposed to infection (Coura & Dias 2009). Transmission to humans occurs primarily through blood-sucking reduviid bugs, but it may also occur through blood transfusion organ transplant, transplacental transmission and oral infection (Moncayo 2003, Dias et al. 2008, WHO 2004, Yoshida 2009). The disease is characterised by an initial acute phase and it is generally accepted that patients with a more severe acute infection may develop a more aggressive chronic phase (Higushi et al. 2003, Coura 2007). Acute diffuse myocarditis is associated with parasite nests and cellular inflammatory foci that are composed of macrophages (Andrade 1991), CD4+ and mainly CD8+ T cells (Henriques-Pons et al. 2002), differentiated as activated/memory T cells (CD62LLow, LFA-1High and VLA-4High) (dos Santos et al. 2001). The chronic phase starts with a usually long-lasting indeterminate period, followed by a symptomatic dilated myocardiopathy in 25-30% of patients. The cytotoxic pathway(s) that destroy cardiomyocytes and the precise correlation between inflammatory response and heart failure are not known. It is important to evaluate which mechanisms regulate cardiac inflammatory processes and their interplay with possible causes of death from this disease. In our previous work, we observed that Fas-L-/- mice (gld/gld) have a very modest cardiac inflammatory response when compared to infected BALB/c. Accordingly, we observed less cardiomyocyte death in gld/gld, yet high mortality rates caused by unknown reasons (de Oliveira et al. 2007). Fas-L, a type II membrane homotrimeric protein, belongs to the tumour necrosis factor family and triggers apoptosis through Fas engagement (Suda et al. 1995). However, Fas activation is also involved in the secretion of cytokines and chemokines, chemotaxis, genomic transcription, cellular activation and other responses (Lambert et al. 2003). Concerning the infection, it has been shown that Fas/Fas-L controls CD4+ T cell population through AICD (activation induced cell death) (Lopes et al. 1999), parasite replication in vitro (Freire de Lima et al. 2000), NO production (Martins et al. 2001) and host cytokine response to T. cruzi infection in vivo, preventing an exacerbated Th2-biased immune response (Lopes et al. 1999, Guilhermo et al. 2007). In other pathologies, such as Coxsackievirus B3, Fas/Fas-L plays a central role in the control of myocarditis, leading cardiac CD4+Th2 (IL-4+) protective cells to death through the cytotoxic activity of σ/δ T cells (Huber et al. 2002, Seko et al. 2002). Various studies associate Fas/Fas-L with renal insufficiency or failure, possibly through apoptotic events not yet completely understood. The most accepted hypothesis for tubular atrophy is based on ischemia induced by apoptosis of tubular epithelial cells mainly through Fas/Fas-L interaction. Thus, in chronic renal illness, this ischemic injury promotes intense necrosis of many cell types (Bohle et al. 1996, Schelling & Cleveland 1999). However, it is well known that human patients with chronic renal failure normally present with anaemia and congestive cardiac insufficiency. This triad named "cardio/anaemic/renal syndrome" (Silverberg et al. 2003) creates a vicious circle, where various substances, such as angiotensin II, endothelin, reactive oxygen species (ROS), epinephrine, erythropoietin and tumour necrosis factor, produced by each checkpoint, can contribute to renal and cardiac failure (Silverberg et al. 2003, Palazzuoli et al. 2008). Based on our previously observed participation of Fas-L in the regulation of acute myocarditis promoted by T. cruzi, the main goal of the present study was to evaluate the influence of this molecule in the death of mice (gld/gld and BALB/c), targeting more systemic possible dysfunctions. We observed similar disturbances in both mouse lineages and concluded that Fas-L is not a central molecule in the regulation of a cardio/anaemic/renal syndrome. However, the lack of Fas-L promotes more severe renal lesions, worsening the course of the infection and probably leading glg/gld to death despite moderate myocarditis. MATERIALS AND METHODS Mice - Seven-week old specific pathogen-free male gld/gld mice and their counterpart isogenic BALB/c were obtained from the Fiocruz animal facility. Mice were housed for at least one week before parasite infection at the Animal Experimentation Division of the Cellular Biology Laboratory-Fiocruz under environmental factors and sanitation conforming to the Guide for the Care and Use of Laboratory Animals [DHEW Publication (NIH) 80-23, revised 1985]. This project has protocol 0099/01 at the Fiocruz Committee of Ethical in Research, according to resolution 196/96 of the National Health Council of the Brazilian Ministry of Health. The number of animals used in each experiment is depicted in figure legends. Parasites and infection - T. cruzi Y strain was maintained by in vivo passages in non-syngeneic Swiss Webster mice and blood trypomastigote forms were isolated as previously described (Araújo-Jorge 1989). Parasites were then diluted in saline and counted in a haemocytometer to adjust the inocula to 1 x 103 parasites/200 μL for intraperitoneal (i.p.) injection. Some dosages and analysis were done on alternate days post infection (dpi), as indicated in graphs, to avoid excessive manipulation of mice. Histopathological analysis - Mice were euthanised on dpi zero, 8 and 15 to collect hepatic and renal tissues to be processed as described elsewhere (Andrade 1990). Briefly, fragments were fixed using Millonnig-Rosman solution (Araújo-Jorge 2000) and 5 μm-thick slices of paraffin embedded samples were further processed and stained with H&E. Qualitative analysis of tissues was based on general appearance and tissue architecture, parenchyma and endothelial cell integrity, glomerular and tubular preservation, edema, cellular inflammatory infiltration and parasite nests. Anti-IgM labelling was carried out using renal tissue slices of cryopreserved TissueTec-embedded (Sakura, CA, USA) kidneys. Endogenous peroxidase activity was blocked with a solution of 3% hydrogen peroxide in phosphate buffered saline (PBS - Sigma) for 5 min. Sections were then rinsed for 20 min in PBS and incubated for 20 min with inactivated normal sheep serum (10% in PBS) to block Fcγ receptors. To identify glomerular IgM deposition, we used a peroxidase-conjugated rat IgG anti mouse IgM mAb (Southern, Birmingham, AL) incubated for 3 h at RT in a moist chamber. Slides were then rinsed with PBS for 15 min and enzyme activity was detected with 3,3'-diaminobenzidine in chromogen solution (Dako Cytomation, Denmark) and counterstained with Mayer's haematoxylin for 1 min. Non invasive parameters Urinalysis - Before the collection of urine samples, mice were adapted for one week (2 h/day) to individual metabolic boxes for sample collection. The urine from control and infected mice was collected on dpi zero, 6, 15 and 20 and immediately analyzed for bilirubin, urobilinogen, ketones, glucose, total protein, blood, nitrite, specific gravity (density), pH and leukocytes (Kit for reacting band system for semi quantitative determination - Uriquest® - Lab Test Diagnóstica, Rio de Janeiro - Brazil). In addition, the time lag for the first urination and urine volume were individually registered. Blood analysis - Serum levels of urea (URE) and CREA (CREA) - We evaluated serum levels of URE and CREA as indicators of renal function in blood samples collected from tail snips on dpi zero, 6, 15 and 20. Ten microliters of serum were collected and diluted in 1 mL of bovine serum albumin 7% to evaluate each parameter, in accordance with the manufacturer, using VITROS® 750XRC Chemistry Analyzer (Ortho-Clinical Diagnostics - Rochester, NY). Blood pressure - Before evaluation of blood pressure, mice were adapted daily for seven days and a tail sphygmomanometer was fitted for three consecutive readings until stabilisation. Blood pressure was individually recorded on dpi zero, 6, 9 and 15 using an LE 5001 Pressure meter® (PanLab Instruments, Barcelona - Spain), evaluating caudal artery pressure in non-sedated animals. Values of systolic, diastolic (DP) and the mean pressure were calculated as indicated by the manufacturer. Echocardiographic studies - Echocardiograph analyses were performed using a 2-dimensional echo Doppler cardiogram (Esaote, model CarisPlus, Firenze-Italy). The standard parasternal long and short axis views were obtained with a 10 MHz transducer and all animals were i.p. tranquilised with diazepan (20 mg/kg body weight) (Compaz® - Cristália Inc, Rio de Janeiro - Brazil). The following parameters were acquired according to the American Society of Echocardiography: diameter of the left ventricle in diastole and systole, septal and posterior wall thickness, shortening fraction (SF), left ventricular ejection fraction and aortic valve area (AVA) at M-mode and B-mode. Isovolumetric relaxation time, peak early (E) and late DP transmitral flow velocities, E deceleration time, ejection time (ET), maximal aortic velocity and flow velocity integral of aortic flow (FVI) were obtained using Echo Doppler. Cardiac output (CO) was obtained from the product of the FVI by AVA. Electrocardiographic studies - All mice were i.p. tranquilised with diazepan (20 mg/kg) and transducers were carefully placed under the skin in accordance with chosen preferential derivation (DII). Traces were recorded using a digital system (Power Lab 2/20) connected to a bio-amplifier in 2 mV for 1s (PanLab Instruments). Filters were standardised between 0,1 and 100 Hz and traces were analyzed using the Scope software for Windows V3.6.10 (PanLab Instruments). We measured heart rate (beats/min-bpm), duration of the PR, QRS, QT intervals and P wave in ms (millisecond) on dpi zero, 8, 15 and 22. The relationship between the QT interval and RR interval was individually assessed. To obtain physiologically relevant values for the heart rate-corrected QT interval (QTc), in units of time rather than time to a power not equal to 1, the observed RR interval (RR0) was first expressed as a unitless multiple of 100 ms, giving a normalised RR interval (RR100 = RR0/100 ms). Next, the value of the exponent (y) in the formula QT0 = QTc x RRy100 was assessed, where QT0 is the observed QT and both QT and QTc are in milliseconds. Taking the natural logarithm of each side of the formula (QT0) = In (QTc) + yln(RR100), the slope of the linear relationship between the log-transformed QT and RR100 thus defined the exponent to which the RR interval ratio should be raised to correct QT for heart rate (Mitchell et al. 1998). Haematology - Red blood series were evaluated on dpi zero, 6 and 13 by directly counting the number of erythrocytes per mL (red blood count - RBC) using a Neubauer chamber with 20 μL of blood diluted in 4 mL of PBS. For haematocrit determination (globular volume - %), we collected 50 μL of blood using heparinised micro capillaries and centrifuged for 5 min at RT. Data are expressed as percentages and were measured using a specific ruler. Haemoglobin was determined based on free haemoglobin levels using a colorimetric assay as follows. Briefly, 250 μL of the working reagent (Hemoglobina®, Lab Test Diagnóstica, Rio de Janeiro - Brazil) were added to 10 μL of freshly collected blood, then the solution was incubated for 5 min at 37ºC and thereafter the optical density (absorbance) was determined using a spectrophotometer (540 nm) (VersaMax®, Molecular Devices, Sunnyvale - CA) as recommended by the manufacturer. The concentration of haemoglobin globular average (CHGM - %) was calculated according to the formula: haemoglobin dosage/globular volume. Therefore, whenever observed, anaemia was classified as hypochromic or normochromic. Captopril and erythropoietin treatment BALB/c and gld/gld mice received captopril (5 mg/L, Ranbaxy Pharmaceuticals, Rio de Janeiro - Brazil) in their drinking water ad libitum from day -1 of infection until death. The captopril solution was changed three times a week and freshly prepared from powder every time. For erythropoietin treatment, both mouse lineages received human recombinant erythropoietin (Dragon Pharmaceuticals, Vancouver, Canada) diluted in saline (0,9%) at a dose of 1.500 U/Kg (200 μL of i.p. injections 3 times/week). Parasitaemia, expressed as parasites/mL, was evaluated in 5 μL of blood collected from tail snips at the indicated time points (Araújo-Jorge 2000). Statistical analysis - The Mann-Whitney non parametric test was used to compare two sets of data (Software SPSS version 8.0) and p values are indicated in figure legends. RESULTS Renal filtration and damage - As cardiac function may also be affected by liquid retention due to decreased renal filtration, we initially measured the urine volume in acutely infected mice (Fig. 1A, B). Our results showed that control BALB/c mice urinated a greater volume (280 μL) (Fig. 1A) than gld/gld mice (130 μL) before infection and on dpi 6 (Fig. 1B). Regarding urine urobilinogen (Fig. 1C, D), we observed a significant increase in infected BALB/c only on dpi 15, but in gld/gld mice it was already increased on dpi 6. Urine leukocyte evaluation (Fig. 1E, F) suggested renal damage in both lineages, but it was also detected earlier in gld/gld (dpi 6), when compared to BALB/c (dpi 15). Other parameters such as bilirubin, ketones, glucose, protein, blood, nitrite, specific gravity (density) and pH showed no significant differences between both lineages (data not shown). Renal function - Renal function was evaluated by measuring URE, CREA, Na+ and K+ in blood and both lineages showed similar levels of all parameters before infection (Fig. 2), indicating no relevant natural renal dysfunction in gld/gld. However, BALB/c and gld/gld showed significantly higher levels of URE from dpi 6 on, although gld/gld was more affected (Fig. 2A, B). As with URE, serum levels of CREA increased after dpi 6, but only at this time point were the levels higher in gld/gld (Fig. 2C, D). Regarding blood electrolytes (Na+ and K+), no significant differences were observed between both lineages (data nor shown). Renal damage - Histopathological analysis of control mice from both lineages showed preserved renal tissue (Fig. 3A, B). However, BALB/c mice on dpi 6 showed discrete damage of proximal tubules and glomerular atrophy (Fig. 3C). Gld/gld mice showed similar alterations, but with a more pronounced outcome associated with a modest inflammatory infiltration (Fig. 3D). On dpi 15, BALB/c mice presented with glomerular atrophy and apparent necrosis, with discrete glomerular haemorrhage and cellular degeneration near proximal tubules (Fig. 3E). Interestingly, gld/gld mice presented with an intense cellular inflammatory infiltration, necrotic degeneration (Fig. 3F), glomerular atrophy and haemorrhage (Fig. 3F, detail). We observed no parasite nests in either group (data not shown). Cardiovascular function - Both lineages presented similar values of mean arterial pressure before infection, but on dpi 6 BALB/c (Fig. 4A) and gld/gld mice (Fig. 4B) showed a significant decrease in blood pressure (hypotension peak). On the other hand, on dpi 15 the arterial pressure increased to above basal levels in BALB/c, whereas this happened from dpi 9 on in gld/gld, with a hypertension peak on dpi 9 (Fig. 4B). Among all parameters evaluated by echocardiography (Table I) (Materials and Methods), no alterations were observed in control mice from either lineage, indicating no relevant natural cardiac dysfunction in gld/gld. However, on dpi 7 infected BALB/c showed abnormal cardiac function, as ascertained by altered values of LV(s), septum (s), ejection fraction, SF and ejection time. In addition, on dpi 14 these parameters had worsened and there were alterations in LV (d), left atrium and Ao/AE (relation between left atrium and aortic base), indicating the decline of cardiac function (Table I). Gld/gld showed basically the same pattern of cardiac alterations, although apparently more severe and established earlier when compared to BALB/c, especially regarding LV (d) and (s) and ET parameters (Table I). Cardiac electric conduction system - Once more we found comparable and normal data regarding cardiac function in both lineages before infection, now based on ECG analysis (Table II). Besides this, a greater difference was observed in the heart rate, which was reduced until dpi 22 in both lineages, but again the alteration was earlier and more pronounced in gld/gld (Table II). Only the QTc interval showed a significant increase on dpi 15 in BALB/c mice and on dpi 22 in both lineages and in gld/gld we detected a sinusal bradycardia and discrete sinusal arrhythmia (data not shown). Red cell evaluation - There was a significant decrease in RBC, globular volume and haemoglobin levels on dpi 13 in both groups (Fig. 5A-F) (murine reference values: RBC = 8.7 x 106/mL, Hct = 44%, Haemoglobin = 12.2 gm/dL) (27). Besides this, CHGM was around 23% in both lineages on dpi 13 (standard value 25.3%) (data not shown) (Fox et al. 1984). These haematological disturbances, hypochromic and normocitic anaemia, could also be playing a role in the cardiac insufficiency and maybe worsening the multiple and systemic alterations that were observed, especially in gld/gld. Moreover, hypochromic and normocitic anaemia are usually associated with renal deficit (Silverberg et al. 2005). Renal lesions and cardiac failure - We evaluated whether IgM deposits could play a role in renal damage and, therefore, in renal dysfunction. We observed no relevant IgM labelling in control (Fig. 6A) or infected BALB/c mice on dpi 6 (Fig. 6B). However, on dpi 15 we observed tissue labelling apparently in the mesangial area (Fig. 6C). Before infection, gld/gld mice presented moderate glomerular IgM deposits (Fig. 6D) and this is possibly a result of many pre-existing immune alterations observed in these mice, such as high levels of auto-immune Igs (Boes et al. 2000). This precipitation increased and a higher glomerular labelling of IgM was observed very early in infected gld/gld mice (dpi 6) (Fig. 6E). However, there was a decrease in the labelling on dpi 15 (Fig. 6F). To evaluate the relevance of angiotensin converting enzyme (ACE) and cardiac function on mortality, BALB/c and gld/gld mice were treated daily with captopril, starting before infection (day-1) as a potential preventive treatment for cardiac and renal dysfunction. Captopril binds to the ACE and inhibits ACE's catalytic production of angiotensin II (Leon et al. 2003). Captopril had no effect on parasitaemia of BALB/c or gld/gld mice (Fig. 6G). However, this treatment postponed and reduced BALB/c mortality and had no influence on gld/gld death (Fig. 6H). DISCUSSION Previous results from our group showed that in the absence of the perforin-dependent cytotoxic pathways, the severity of acute myocarditis is increased after T. cruzi infection (Henriques-Pons et al. 2002). We therefore decided to examine whether the Fas-L-dependent pathway would also influence the evolution of myocarditis after infection and observed that in gld/gld mice there is a very mild cardiac inflammation, but a high mortality rate (de Oliveira et al. 2007). There are few studies addressing the Fas-L relevance in T. cruzi infection, such as in: (i) atrophy of mesenteric lymph nodes (de Meis et al. 2006) and thymus (Henriques-Pons et al. 2004), (ii) NO production and apoptosis in splenocytes (Martins et al. 1999) and (iii) control of Th1/Th2 balance (Lopes et al. 1999). Recent publications report that a Th2-biased immune response with higher levels of IL-10 and IL-4 in gld/gld is associated with increased susceptibility to experimental infection (Guilhermo et al. 2007), although it might be different in human patients (Araújo et al. 2007). It is possible that T. cruzi-infected gld/gld mice are more susceptible to the infection due to a complex array of pre-existing and infection-imposed systemic alterations, including components of the immune system and organ dysfunction. In agreement with our results regarding myocarditis in infected gld/gld mice, it was published that coxsackievirus B3-induced myocarditis is also decreased in the absence of Fas/Fas-L interaction (Seko et al. 2002). However, after T. cruzi infection all gld/gld died in the early acute phase, suggesting that other disturbances can occur and lead gld/gld to death. Thus, the goal of the present study was to investigate the participation of FasL in the possible systemic alterations, other than myocarditis, that could be related to the death of T. cruzi infected mice. To date, human patients with anaemia and chronic renal failure have more severe cardiac insufficiency (Silverberg et al. 2003). Moreover, anaemia is found in about one third of all cases of congestive heart failure (CHF) and is the most likely common cause of chronic renal insufficiency, which is present in about half of all CHF cases (Silverberg et al. 2004). The authors considered and discussed the existence of a complex feedback known as cardio/anaemic/renal syndrome (Silverberg et al. 2003). In the present paper we observed a decrease in haemoglobin concentration on dpi 6 and this anaemia could therefore aggravate renal damage and cardiac insufficiency during infection in both lineages, although worsened in gld/gld. Therefore, we treated in vivo with recombinant erythropoietin, since it has been used in the treatment of anaemia associated with renal failure, leading to the improvement of cardiac function (Silverberg et al. 2005). However, we observed no reversion in the mortality rate of either mouse lineage after treatment (unpublished observations), thus suggesting that anaemia does not directly lead mice to death, but it may play a role when combined with other dysfunctions. Our results showed an increase in URE and CREA, oliguria and even anuria in mice from both lineages, clearly indicating renal dysfunction (Nelson & Couto 1992). Moreover, histopathological analysis showed glomerular IgM deposits (we found no IgG precipitates) (data not shown), possibly leading to complement activation, renal damage and acute kidney injury (AKI) (Thadhani et al. 1996). However, additional pathophysiological alterations could be taking place, such as compensatory neuronal-endocrine mechanisms, leading to tissue damage. We do not know why infected gld/gld have earlier renal IgM deposits, but these deposits may be induced by a combination of pre-existing alterations of the innate immune system of these mice and the infection, favouring the precipitation of polyreactive IgM molecules on dpi 6. Moreover, we observed degeneration of proximal tubules, perivascular edema and interstitial congestion in both groups of mice. On dpi 15 we observed the aggravation of tubular damage in gld/gld and intense inflammatory infiltration. AKI may have several causes: inflammatory infiltration, endothelial damage, ROS increase, production of nitrogen species and other mediators of tubular cells' sub-lethal or lethal damage (Boneventre & Zuka 2004, Friedewald & Rabb 2004). After necrotic death, many pro-inflammatory mediators are released, culminating in the recruitment of blood leukocytes and cellular activation/function (Cines et al. 1988). Fas/Fas-L interaction plays an important role in these steps (Kataoka et al. 2000, Thone & Tschopp 2001) and also in apoptosis. We do not know yet why gld/gld mice have a higher renal inflammatory response but more moderate myocarditis, when compared to BALB/c (de Oliveira et al. 2007). Fewer cells migrate to the cardiac tissue of gld/gld, but in both lineages we found a cardiac CD4/CD8 double negative T cell population. However, only these cells collected from gld/gld had a phenotype that suggested attenuation of cellular effector functions (de Oliveira et al. 2007). These findings illustrate the complexity of the roles played by Fas/Fas-L in the regulation of the inflammatory response in different organs during infection. On the other hand, our data regarding a cardio/anaemic/renal syndrome indicate that both lineages have similar physiological and general alterations, but with an earlier and worse outcome in gld/gld. For example, arterial hypotension is observed on dpi 6 in both infected lineages, with high levels of CREA. Hypotension and shock are common events in critically ill patients, leading to AKI and renal failure that may promote accumulation of toxic substances, vasoplegia and finally hypotension. Moreover, moderate hyperchloraemic acidosis may promote vasodilatation and thus lower blood pressure, impairment of CO and decreased perfusion in both liver and kidneys, probably through down-regulation of Beta-2 receptors (Hoste & Kellun 2006). We observed very early signs of cardiac insufficiency, such as a reduction of contraction capacity and ejection fraction and it is reasonable to hypothesise that renal damage is involved in heart failure through the activation of the renin-angiotensin system (RAS), for example. Most of the well-known cardiovascular and renal effects of RAS are attributed to ACE, but less is known about its function in the cardiac muscle itself. Functional angiotensin II receptors have been documented in cardiac fibroblasts as well as an intracardiac effect induced by short and long-term aldosterone stimulus (Linjnem & Petrov 2003). In vitro, angiotensin II increases cardiac fibroblast-mediated collagen synthesis and mRNA levels of collagen type I, type III, pro-alpha1 (I) collagen, pro-alpha1 (III) collagen and fibronectin, as well as inhibited matrix metalloproteinase I activity. The angiotensin II-dependent secretion and expression of collagen was completely abolished by AT1 receptor antagonism, but not by AT2 antagonists (Linjnem & Petrov 2003). Moreover, in kidneys, ACE2 protein levels were significantly decreased in hypertensive rats, suggesting a negative regulatory role of ACE2 in blood pressure control (Danylczyk & Penninger 2006). We observed in the present paper that captopril treatment postponed BALB/c mortality but had no effect on gld/gld death. This may be interpreted by at least two not mutually exclusive alternatives: (i) pre-existing alterations predisposed gld/gld mice to very early and important renal damage that was not sufficiently reverted by captopril and/or (ii) although BALB/c and gld/gld apparently present the same sequence of central events, certain checkpoints may be more directly related to death in either lineage. Beyond the renal target of ACE activity, captopril acts to a certain extent reducing systemic arterial pressure, peripheral vascular resistance, cardiac filling pressure and increasing CO. Although captopril is also an anti-inflammatory agent, no alteration in T-cell proliferative response to T. cruzi or in the levels of T. cruzi and myosin-specific IgG was observed (Leon et al. 2003). Moreover, captopril upregulates bradykinin, leading to nitric oxide synthesis (Anning et al. 1997, Gallagher et al. 1998), which may be important in the resistance to acute T cruzi-induced myocarditis. It is also important to observe that captopril reduces cardiac necrosis and fibrosis in T. cruzi infected mice (Danylczyk & Penninger 2006). In summary, mice infected with T. cruzi develop a cardio/anaemic/renal syndrome in the acute phase and our observations indicate antagonistic Fas-L-based immunoregulatory functions in heart and kidney. In the absence of Fas-L, there is minor myocarditis but a higher renal inflammatory response and damage (de Oliveira et al. 2007), possibly due to earlier renal IgM deposits. This renal dysfunction would, in turn, increase negative effects induced by the infection in the cardiovascular system, affecting blood pressure and cardiac dysfunction, for example. Considering the anaemia, these alterations would lead to a cycle that could be responsible for the death of the mice. However, although BALB/c mice experience similar cardiac/renal alterations (damage/insufficiency), these events occur later in the infection, are less intense and can be partially reverted by captopril treatment. ACKNOWLEDGEMENTS To Mr. Luis Lopes Carvalho, for invaluable assistance in the pathological analysis, and to Dr. Alejandro Marcel Hasslocher Moreno, for important suggestions. REFERENCES
The following images related to this document are available:Photo images[oc09220f3.jpg] [oc09220f1.jpg] [oc09220f2.jpg] [oc09220f6.jpg] [oc09220t2.jpg] [oc09220f5.jpg] [oc09220t1.jpg] [oc09220f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}