 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 104, No. 8, 2009, pp. 1100-1110 ARTICLES A new approach for potential drug target discovery through in silico metabolic pathway analysis using Trypanosoma cruzi genome information Marcelo Alves-FerreiraI; Ana Carolina Ramos GuimarãesI; Priscila Vanessa da Silva Zabala CaprilesII; Laurent E DardenneII; Wim M DegraveI, + ILaboratório
de Genômica Funcional e Bioinformática, Instituto Oswaldo Cruz-Fiocruz,
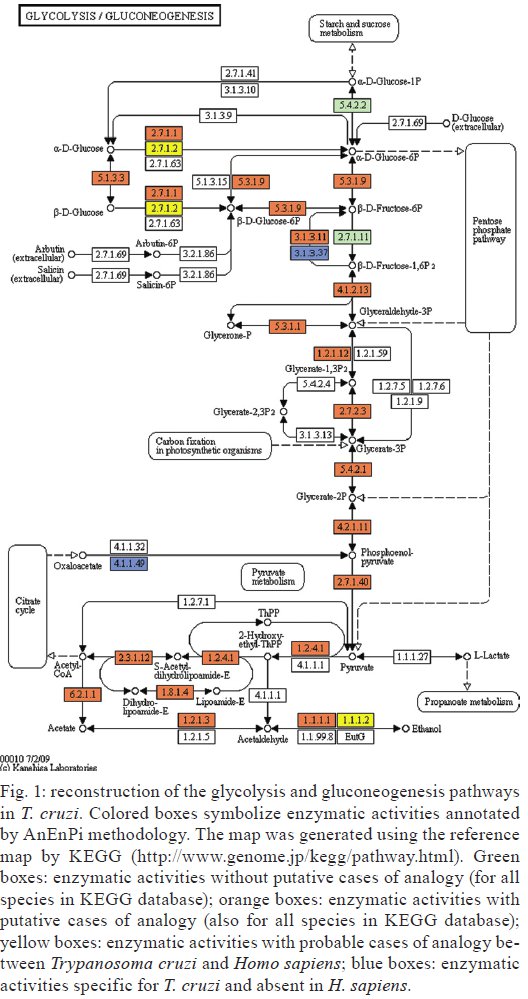
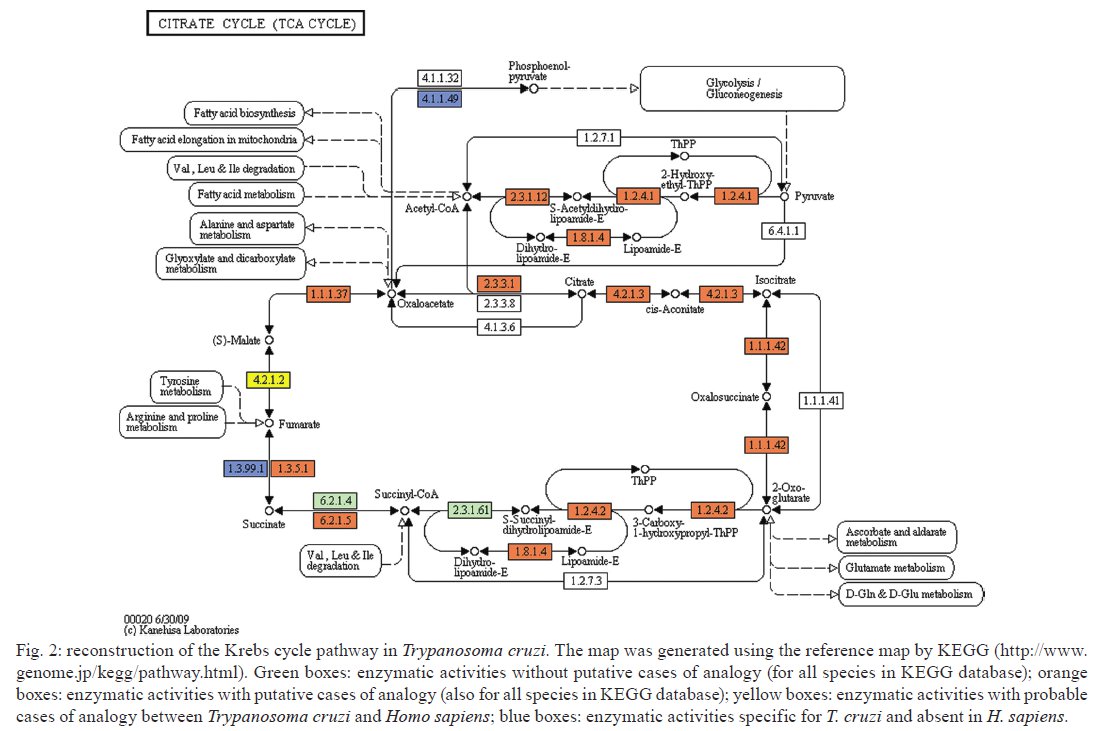
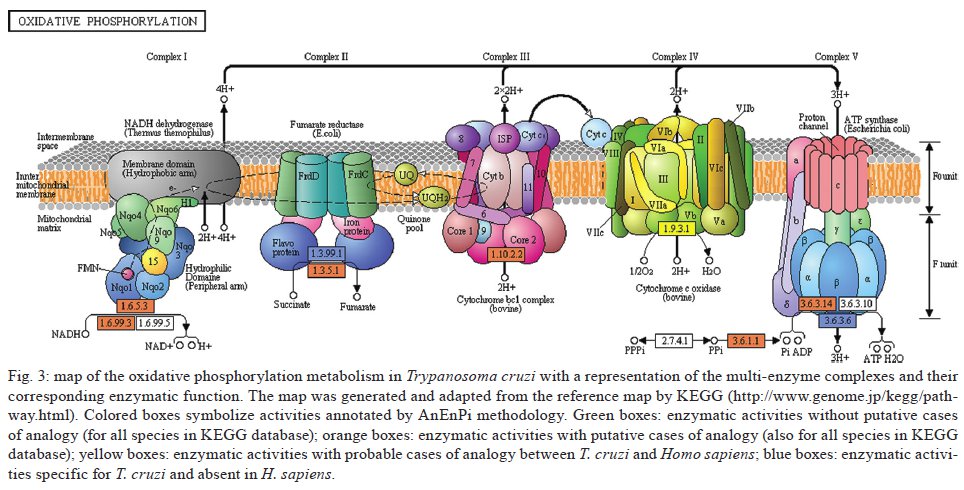
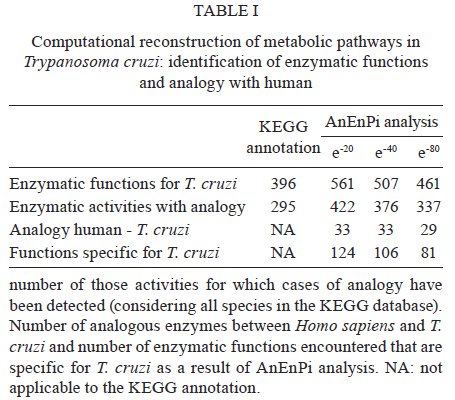
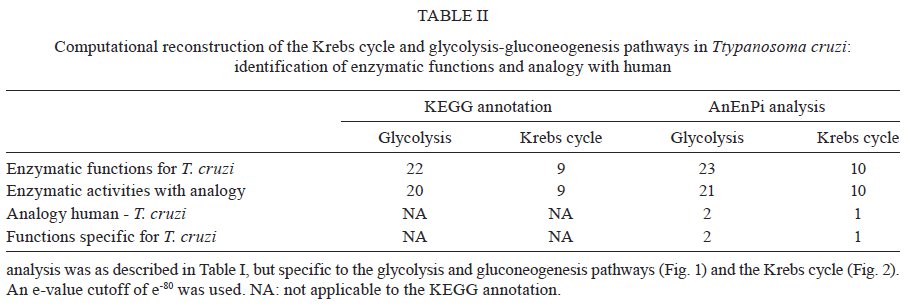
Av. Brasil 4365, 21045-900 Rio de Janeiro, RJ, Brasil Received 10 June 2009 Code Number: oc09224 ABSTRACT The current drug options for the treatment of chronic Chagas disease have not been sufficient and high hopes have been placed on the use of genomic data from the human parasite Trypanosoma cruzi to identify new drug targets and develop appropriate treatments for both acute and chronic Chagas disease. However, the lack of a complete assembly of the genomic sequence and the presence of many predicted proteins with unknown or unsure functions has hampered our complete view of the parasite's metabolic pathways. Moreover, pinpointing new drug targets has proven to be more complex than anticipated and has revealed large holes in our understanding of metabolic pathways and their integrated regulation, not only for this parasite, but for many other similar pathogens. Using an in silicocomparative study on pathway annotation and searching for analogous and specific enzymes, we have been able to predict a considerable number of additional enzymatic functions in T. cruzi. Here we focus on the energetic pathways, such as glycolysis, the pentose phosphate shunt, the Krebs cycle and lipid metabolism. We point out many enzymes that are analogous to those of the human host, which could be potential new therapeutic targets. Key words: Trypanosoma cruzi - metabolism - metabolic pathways - drug target - analogous enzyme Chagas disease, which is caused by an infection with the kinetoplastid protozoan parasite Trypanosoma cruzi, remains a serious public health problem in most Latin American countries despite successes in controlling transmission through vector control and blood donor screening in the affected regions. It is estimated that about 12 million people are chronically infected with Chagas disease (Dias 2006). In addition, due to migration, Chagas disease has become a threat in several additional countries (Schmunis 2007). The acute phase, which appears shortly after infection, is often hard to notice and after a silent period of a several years or decades a chronic phase develops in about one-third of the infected individuals. Due to this clinical evolution, Chagas disease is often considered a "silent killer," impairing early specific diagnosis and treatment (Tarleton 2007, Bilate & Cunha-Neto 2008). The main clinical manifestations of Chagas disease are mostly cardiac, but also digestive alterations and the pathogenesis is attributed to host immune system disturbances associated with a very low and often undetectable parasite presence (Rocha et al. 2007, Marin-Neto et al. 2008). Until now, there has been no immediate prospect for the development of a vaccine and current clinical therapy based on heterocyclic nitro compounds (nifurtimox and benznidazol) is quite unsatisfactory for chronic patients, thus calling attention to the search for new therapeutic approaches (Coura & Castro 2002, Dias 2006). The different developmental stages and changing biochemical interactions between the parasite and the vertebrate host during the life cycle of T. cruzi render the development of new drugs more difficult (de Souza 2002). The availability of genomic (El-Sayed et al. 2005) and proteomic data (Atwood et al. 2005) from the parasite has set high hopes for the identification of new drug targets and a large amount of data is currently accessible (http://tcruzidb.org/tcruzidb/, http://www.genedb.org/, http://tritrypdb.org/tritrypdb/ and http://eupathdb.org/eupathdb/). However, difficulties with whole genome assembly due to the highly repetitive nature of T. cruzi sequence content, together with the heterozygosity of the analysed strain and the presence of many predicted proteins with unknown or unsure functions has hampered a full view of the metabolic pathways in this parasite. A lack of insight into the regulation and interplay of those pathways, the limited tools for functional genetic analysis to prove the essentiality of certain enzymes and little knowledge on functional differences with corresponding human enzymes are causes for the slow development of new drug candidates. Moreover, drug development for neglected diseases is not widely met with the necessary attention and determination that the many patients and people at risk deserve, which has been largely discussed on different occasions. In eukaryotes, the metabolism can be organised in pathways that are well conserved between different phyla; however, a considerable amount of variability can be noted in enzymatic characteristics, pathways, cellular compartments and regulatory mechanisms when species are compared. Protozoa, like the Kinetoplastida, present a number of features that are distinct from those seen in other organisms, including several organelles that are absent in mammalian cells (de Souza 2005, de Souza et al. 2008). One such organelle, the acidocalcisome, was discovered more recently in trypanosomatids and in the Apicomplexa (Docampo et al. 2005). The organelle has an important role in the regulation of calcium, phosphor and other elements though the presentation of several families of transporters on the membrane. The glycosome, which is a special type of peroxysome, is typical of trypanosomatids and contains seven enzymes of the glycolytic pathway; enzymes involved in carbohydrate metabolism, lipid metabolism (beta oxidation of fatty acids and their elongation, synthesis of lipid ethers and some steps of isoprenoid synthesis) and metabolism of purines and pyrimidines; and is an important generator of redox potential in the form of NADPH (Michels et al. 2000, Moyersoen et al. 2004). The mitochondria, vital organelles for the respiratory process and ATP production, participate in several metabolic cycles and are related to the process of cell death. Recent studies have indicated that the mitochondrial membrane of trypanosomatids has a peculiar lipid composition. The specific replication and editing processes, associated with the already well studied unusual DNA structure of this organelle, represent potential targets for new drugs. The trypanosomatids have important and fundamental differences in their processes to generate energy, although some pathways are common (Tielens & Van Hellemond 1998). The parasitic forms of trypanosomatids also have metabolic differences during their life cycle, such as the Krebs cycle in Trypanosoma brucei, which is nearly inactive in bloodstream forms found in the vertebrate host, but is active in the procyclic form in the invertebrate host (Hannaert et al. 2003). Even so, some of the glycosome enzymes are involved in amino acid metabolism, others in fatty acid synthesis and two enzymes participate in gluconeogenesis (van Weelden et al. 2005). All these particularities constitute potential new drug targets. Over the last 100 years, different methods and drugs have been used against T. cruzi infection (Coura & Castro 2002). Since the 60s, nitrofurans, such as nifurtimox (LampitTM) and later imidazole compounds, such as benznidazole (RochaganTM), have been used due to their oxidising action, through the formation of reactive radicals and their ability to block enzymes and lipids. Newer chemicals were developed based on the nitrofuran and nitroimidazole backbone in an attempt to augment activity while diminishing the many negative side effects of the drugs. These drugs, which include ketoconazole, fluconazole, itraconazole and posaconazole, showed higher efficiency against the parasite and were also shown to inhibit enzymes in steroid biosynthesis (Urbina & Docampo 2003, Liñares et al. 2006). These triazole (antifungal) agents are known to block the synthesis of ergosterol through the inhibition of the enzyme cytochrome P450 lanosterol 14a-demethylase, leading to the accumulation of methylated sterol precursors. Inhibitors of nucleotide metabolism, such as the xantine oxidase inhibitor alopurinol, were also evaluated (Maya et al. 2007). The trypanosomatid specific enzyme tripanothione reductase has been extensively analysed as a potential drug target and both chemical compounds (Maya et al. 2007, Perez-Pineiro et al. 2009) and natural products (Galarreta et al. 2008) were found to inhibit the enzyme. Cruzipain, a cysteine proteinase involved in cellular processes and host-parasite interactions, was pointed out as another potential target and in vivo assays with different chemical inhibitors were quite promising (Doyle et al. 2007, Brak et al. 2008, Fricker et al. 2008). Several other enzymes have also been targeted, such as gliceraldehyde-3-phosphate dehydrogenase (Freitas et al. 2009), pteridine reductase (Cavazzuti et al. 2008), dihydrofolate reductase/thymidilate synthase, farnesyl-pyrophosphate synthase and DNA topoisomerase (Paulino et al. 2005), while a large number of natural products are also being screened. These studies seem quite promising and a decisive breakthrough is eagerly awaited from the scientific community. A potential new drug target should be a molecule that is essential to the parasite, for which high affinity inhibitors can be identified and which have little or no effect on the eventual human homologue. Specificity, low toxicity, bioavailability, stability and ease of administration are additional requirements for potential drug candidates. Strategies usually aim to identify parasite specific enzymes, complete biochemical pathways (Karp et al. 1999) or structural differences between human and parasite homologues. The latter approach is hampered by the limited availability of 3D structures. With this in mind, we developed a methodology to identify cases of enzyme analogy, where two different proteins share the same function or activity while lacking similarity between their primary sequences (Galperin et al. 1998). In these cases, the enzymes are thought to have originated through independent evolution. Therefore, we designed a computational workflow and tool, AnEnPi (Otto et al. 2008), to detect them from genomic data. The absence of (detectable) primary sequence similarity allows for the expectation of a substantially different folding of the protein. Besides the importance of identifying analogous enzymes in parasites and humans as potential drug targets, the identification of structurally unrelated enzymes sharing the same enzymatic activity may reveal new catalytic mechanisms and shed light on the origin and evolution of biochemical systems and pathways. Previously, we analysed the amino acid metabolic pathways of T. cruzi (Guimarães et al. 2008). Here we further explore the metabolic pathways of T. cruzi to assess the extent of analogy in the parasite, while suggesting important new candidates for drug targeting. MATERIALS AND METHODS AnEnPi analysis of the metabolic pathways of T. cruzi was essentially done as previously described (Otto et al. 2008). Briefly, the AnEnPi tool was used to cluster a dataset composed of 311 reference metabolic pathways and 1,871,732 protein sequences of 36 eukaryotes, 398 eubacteria and 31 archaeabacteria obtained from the KEGG database (Kanehisa et al. 2006). In total, 326,013 sequences had a corresponding EC number assigned to them describing their enzymatic activity, which belong to 2,433 different EC classes. This result forms the main dataset for clustering used by AnEnPi. Sequences with less than 100 amino acids were excluded and an all-against-all BLASTp was performed for each enzymatic activity. Enzymes were considered to be homologous if they belonged to the same EC class and had a similarity score greater than or equal to 120 (corresponding to an e-value close to 10-6) (Galperin et al. 1998). Enzymes in the same EC class with very low or undetectable similarity clustered in separate groups and were considered analogous. The resulting dataset was further refined by excluding all clusters with only one sequence (singlets) or where the enzymatic activity was not defined up to the fourth level of EC classification. Furthermore, all clusters of a determined function, with proteins annotated as subunits of this function and belonging to the same species, were joined. The results can be visualised as a metabolic map through the use of an external (KEGG) resource. For the purpose of this analysis, a potential case of analogy was identified if the sequences from a given enzymatic activity, which were present in the genome of a single organism or between two organisms, were placed in different clusters after grouping (intra-genomic and inter-genomic analogy, respectively). Using this method, orthologs and paralogs would end up in the same cluster. AnEnPi maps the presence of a given function, within a single species or between two species, in one or more clusters. For metabolic analysis, we used the T. cruzi genomic TSK-TSC v5.0 dataset of 19,607 predicted proteins (http://tcruzidb.org). Tentative function was annotated using BLASTp with e-value cut-offs of 10-20, 10-40 or 10-80 and subsequently assigned to clusters of enzyme activities derived from the KEGG database as explained above. In the current study, analogous enzymes in the parasite were identified in comparison with enzymes in the corresponding human metabolic pathways as exemplified in Figs 1 , 2 and 3. RESULTS AND DISCUSSION Many aspects of T. cruzi biochemistry have been investigated and described over the last few decades and several remarkable features can be noted in the major metabolic pathways of the parasite, which may constitute valuable focal points for further research and drug development. Using AnEnPi, we performed a detailed analysis of the enzymes predicted from the T. cruzi CL-Brener genome sequence (as available from http://tcruzidb.org). Using this method we were able to tentatively annotate a considerable amount of additional enzymatic functions for the parasite, compared to the KEGG prediction, as shown in Table I. Using e-value cut-offs of 10-20, 10-40 and 10-80 we found 165, 111 and 65 additional protein sequences, respectively, with probable enzymatic functions, compared to the 396 functions assigned for T. cruzi using KEGG. For the subset of activities present in the parasite, 74% (295) had cases of analogy in KEGG displaying more than one cluster, as opposed to 40.5% when analysing all pathways from all species in the database (Otto et al. 2008). For protein functional annotation using an e-value of 10-20, AnEnPi pointed out 127 additional cases of analogy. When comparing the corresponding human enzymes, AnEnPi could find 33 analogous enzymes in T. cruzi and 124 enzymatic functions that are not present in humans. Table II shows the results of this analysis for the glycolysis and gluconeogenesis pathways and for the Krebs cycle. The corresponding metabolic maps are shown in Figs 1 and 2. Lipid and energy metabolism of the parasite have received considerable attention as potential drug targets in T. cruzi and are briefly reviewed below. Lipid metabolism Lipids have essential roles in biological membranes and are sources of stored energy. They also perform several other functions, such as being cofactors for enzymes, acting as hormones, being intra and extra-cellular signal messengers and involvement in protein anchorage to membranes and transporters. In trypanosomes, several molecules, such as miltefosine and azasterol, have been evaluated as structural analogues of lipids, phospholipids and steroids. They function as inhibitors of key pathways in the synthesis of lipids and steroids, respectively (Croft et al. 2003, Roberts et al. 2003). The lipid metabolism of trypanosomatids has been studied since the 60s (Korn & Greenblatt 1963, Korn et al. 1969, Dixon et al. 1971, 1972) but the identification of key differences between the host and the parasite, as well as the study of the enzymes themselves, has not been fully achieved. The sterol biosynthesis pathway has been better characterised, especially because of its similarity with the corresponding pathways in fungi and yeasts where the main sterol is ergosterol (Roberts et al. 2003). Most of the experimental work is being done in T. brucei because this parasite can be subjected to gene silencing through RNAi (Lee et al. 2006, van Hellemond & Tielens 2006, Stephens et al. 2007). Several enzymes of the pathway have been cloned from T. cruzi and Leishmania and experimental drugs that affect these enzymes are being evaluated (Urbina 1997, Magaraci et al. 2003, Pourshafie et al. 2004, Lorente et al. 2005). The first step in the synthesis of isoprenoid, steroids and other lipid components, such as farnesyl and dolichol, involves the synthesis of isopentenyl-pyrophosphate. There are two pathways for the synthesis of this compound: the mevalonate pathway from the acetyl-CoA precursor and the pathway via DOXP/MEP (1-deoxy-D-xylulose 5-phosphate/2-C-methyl-D-erythritol 4-phosphate), which includes glyceraldehyde-3-phosphate and pyruvate as precursors. The latter is present in algae, some bacteria and in protozoa apicomplexa and is targeted by antiparasitic drugs, such as fosmidomycin and derivates (Wiesner & Jomaa 2007, Haemers et al. 2008). On the other hand, higher eukaryotes and trypanosomatids synthesise isoprenoids through the mevalonate pathway (Urbina 1997, Buhaescu & Izzedine 2007). Careful analysis of each enzyme in the pathway is under way, including the HMG-CoA reductase in several trypanosomatids (Pena-Diaz et al. 1997, Montalvetti et al. 2000, Hurtado-Guerrrero et al. 2002) and the mevalonate kinase in Leishmania major (Sgraja et al. 2007). We are studying two enzymes from this pathway: isopentenyl diphosphate isomerase and phophomevalonate kinase, both of which are analogous to the corresponding human proteins (M Alves-Ferreira, unpublished observations). The steroid metabolism in trypanosomatids was initially studied through the characterisation of intermediate metabolites using isotopic labelling and inhibitors for several yeast enzymes (Roberts et al. 2003). In epimastigotes of T. cruzi, ergosterol and ergosta-5,7,-dien-3β-ol represent about 40% of the steroids, while estigmata-5,7-dien-3β-ol and ergosta-5,7,22-trien-3β-ol represent about 30% (Beach et al. 1986). The amastigote form of the parasite does not seem to express the Δ5 and Δ22 desaturases and 80% of total sterol was cholesterol, which was probably present through incorporation from the host cells (Liendo et al. 1999). The presence of variants with substitutions on carbon-24 and the therapeutic effects of azasterols on the parasites clearly show the importance of the esterol-24-metyltranferase enzyme in the biosynthesis, besides the overall importance of the ergosteroids in the biology of the parasites. The enzyme sterol-24-metyltransferase has been described in several species of Leishmania, in T. cruzi and in T. brucei (Magaraci et al. 2003, Pourshafie et al. 2004, Jiménez-Jiménez et al. 2006). The exact intracellular localisation of the enzyme in T. cruzi is still unclear and was proposed to be associated with the glycosome as well as the cytoplasm. In our analyses using AnEnPi, we identified three homologous gene copies that encoded this enzyme in T. cruzi, corroborating the previous description of this enzyme and its indication as a possible new therapeutic target. The energy metabolism Energy metabolism begins with nutrient uptake. In T. cruzi, a single isoform of the hexose transporter THT1 was described and several copies are present in the genome. In T. brucei, two isoforms of the transporter were identified (Bringaud & Baltz 1993, Tetaud et al. 1994). The transporter in T. cruzi has a high affinity for glucose, but can also transport other monosaccharides, such as D-fructose, which differs from the human transporter GLUT1 (Tetaud et al. 1994, Barrett et al. 1998). More recently, amino-acid transporters were also described for T. cruzi, including high affinity systems for arginine (Pereira et al. 1999) and aspartate (Canepa et al. 2005), as well as high and low affinity transporters for proline (Silber et al. 2005). A glutamate transporter was reported in T. cruzi and it was demonstrated that the uptake of this amino-acid is five times higher in epimastigotes than in metacyclics (Silber et al. 2006). It was further shown that aspartate, glutamine, asparagine, methionine, oxaloacetate and alpha-ketoglutarate also compete for use of this transport system. Glycolysis - The enzymes of the glycolytic pathway in trypanosomatids are organised in two cellular compartments, while in higher eukaryotes these enzymes are present in the cytoplasm. In T. brucei, the first seven enzymes of the pathway are present in the glycosome and transform glucose into 3-phosphoglycerate, while the three other enzymes are localised in the cytoplasm (Hannaert et al. 2003, Bringaud et al. 2006). T. brucei is an exclusively extracellular parasite and its metabolism is directly related to the concentration of nutrients in the medium. The slender bloodstream forms of the parasite produce energy almost exclusively from glucose. The phosphoglycerol kinase localisation is either glycosomal or cytosolic, as observed in the promastigotes of different Leishmania species and in T. cruzi epimastigotes and are required to maintain the glycosomal ATP/ADP balance, which offers a rationale for the presence of both phosphoglycerate mutase and enolase in the cytosol (Hannaert et al. 2003). In anaerobic conditions, T. brucei produces mostly pyruvate and re-oxidises NADH through the use of an alternative oxidase (van Hellemond et al. 2005). However, T. cruzi and Leishmania species require a more complex energy metabolic system and additional pathways, such as the Krebs cycle and oxidative phosphorylation, are compartmentalised in the mitochondria (Taylor & Gutteridge 1987, Hannaert et al. 2003). In T. cruzi, the first enzyme of the pathway, a hexokinase (HK), was described as having different kinetic characteristics than the human enzyme. It did not suffer inhibition by D-glucose-6-phosphate or by other vertebrate HK regulators, such as fructose-1,6-diphosphate, phosphoenolpyruvate, lactate or citrate; although there is a weak competitive inhibition by ADP with respect to ATP (Racagni et al. 1983, Urbina & Crespo 1984). More recently, additional experiments showed that the enzyme is inhibited in a non-competitive way by inorganic pyrophosphate (PPi) and does not phosphorylate other sugars, such as fructose, mannose and galactose (Cáceres et al. 2003). This suggests that biphosphonates are possible inhibitors of this T. cruzi enzyme. Therefore, the synthesis of 42 compounds was described, in which the best of this series demonstrated a Ki of 2.2 μM against amastigotes. Other reports in this direction showed the potent and selective inhibition of HK and inhibition of the proliferation of the clinically relevant intracellular amastigote form of the parasite, using aromatic arinomethylene biphosphonates, which act as non-competitive or mixed inhibitors of HK (Hudock et al. 2006, Sanz-Rodr1guez et al. 2007). However, Cordeiro et al. (2007) showed that the parasite also has a glucokinase with ten times higher affinity for glucose preferentially in a β-D-glucose isomeric form, while the preferential HK substrate is the α-isomer. Using the AnEnPi tool, in silico annotation of T. cruzi predicted proteins indicated the presence of both HK and glucokinase. Fig. 1 shows the reconstruction of the glycolytic pathway and where these two enzymes are located (EC 2.7.1.1 and EC 2.7.1.2). Unlike the HK in this analysis the glucokinase is clearly analogous to the human enzyme and constitutes a possibly interesting therapeutic target in the parasite. Recently, Chambers et al. (2008) showed that the anti-cancer drug ionidamine was capable of inhibiting one of the HKs (TbHK1) from T. brucei and was effective against both the recombinant enzyme as well as the bloodstream and procyclic forms of this parasite. Another point of regulation in the glycolytic pathway of eukaryotes resides in the catalytic step of the phosphofructokinase. This enzyme displays different characteristics in T. cruzi, including the fact that the enzyme is not sensitive to typical modulators, such as the activator fructose-6-P and the inhibitors ATP, citrate and phosphoenolpyruvate (Urbina & Crespo 1984, Adroher et al. 1990). The triose phosphate isomerase (TIM) is also a central enzyme of the glycolytic pathway that has been studied in T. cruzi and in a number of pathogenic protozoa (Pérez-Montfort et al. 1999, Reyes-Vivas et al. 2001, Rodríguez-Romero et al. 2002, Olivares-Illana et al. 2006). Recently, two studies showed the possibility of developing new therapeutic agents against trypanosomes using TIM as a molecular target. The compound 6, 6'-bisbenzothiazole-2, 2' diamine in the low micromolar range was able to specifically inhibit the TIM of trypanosomatids (Olivares-Illana et al. 2006), while the compound dithiodianiline in nanomolar concentrations was able to completely inhibit the recombinant TIM of T. cruzi and was trypanocidal for the epimastigote form of the parasite (Olivares-Illana et al. 2007). The phosphoglycerate kinase (PGK) was described by Concepción et al. (2001) as having two isoforms: one of 56 kDa, which is exclusively glycosomal, and a second of 48kDa, which is expressed both in the cytoplasm and in glycosomes. In the same study, the authors demonstrated that 20% of total PGK activity is found in glycosomes and 80% in the cytoplasm. The pyruvate kinase in T. cruzi was first reported by Juan et al. (1976) and following studies demonstrated the kinetic details, regulation and localisation of this enzyme (Cazzulo et al. 1989, Adroher et al. 1990). The enzyme alcohol dehydrogenase (EC: 1.1.1.2), which is important in oxidoreduction of alcohol, can be considered a possible target for therapeutic studies against T. cruzi because AnEnPi analysis indicates it as being an analogue to the enzyme found in humans (Table II). It was demonstrated that the corresponding enzyme from Entamoeba histolytica can be inhibited by cyclopropyl and cyclobutyl carbinols and is considered as a possible new drug target for this parasite as well as for Giardia lamblia (Espinosa et al. 2004). The pentose phosphate shunt - The pentose phosphate shunt is the most studied pathway in T. cruzi and the participating enzymes have been well characterised both molecularly and biochemically (Igoillo-Esteve et al. 2007). The enzymes of this pathway were identified as cytoplasmic, but with secondary localisation in the glycosome. They play an important role in the generation of NADPH, which is essential in responding to oxidative processes of the host defence system. Maugeri and Cazzulo (2004) showed that under normal conditions 10% of the glucose captured by T. cruzi is metabolised in the pentose pathway. All seven enzymes of the pathway are expressed in the four life stages of T. cruzi (Maugeri & Cazzulo 2004) and this pathway can be divided into oxidative and non-oxidative branches. The oxidative branch is responsible for the production of NADPH and ribose-5-phosphate and is regulated by the ratio of NADP/NADPH through the first enzyme in the process, glucose-6-phosphate dehydrogenase (G6PD). This enzyme has several copies in the T. cruzi genome, while the 6-phosphogluconolactonase, the 6-phosphogluconate dehydrogenase (6PGD) and the ribose 5-phosphate isomerase are present as only one copy. The latter enzyme is analogous to the human enzyme, which makes it an interesting therapeutic target (Igoillo-Esteve et al. 2007). Recently, Stern et al. (2007), using site directed mutagenesis, revealed details on the reaction mechanism of this enzyme, which acts via the formation of the 1,2-cis-enediol. They also showed that the 4-phospho-D-erythronohydroxamic acid, an analogue to the reaction intermediate, competitively inhibits this enzyme with a Ki = 1.2 mM and an IC50 of 0.7 mM. Mielniczki-Pereira et al. (2007) studied the two enzymes responsible for NADPH production, G6PD and 6PGD and showed differences in the activities of those enzymes between the Tulahuen 2 and Y strains, as well as the importance of G6PD in protecting the parasite against reactive oxygen species. The non-oxidative branch complements the pathway and the enzymes involved are ribulose-5-phosphate epimerase, with two gene copies in T. cruzi and the transaldolase and transketolase, each with one copy per haploid genome (Igoillo-Esteve et al. 2007). One of the copies of ribulose-5-phophate isomerase presents a C-terminal (PTS1) signal for glycosome targeting but the cytosol also showed activity (Maugeri & Cazzulo 2004). A transaldolase is responsible for the transfer of the dihydroxyacetone group from fructose-6-phosphate to erythrose-4-phosphate, leading to the synthesis of sedoheptulose-7-phosphate and glyceraldeyde-3-P. The biochemical characterisation of the recombinant enzyme was reported (Igoillo-Esteve et al. 2007). The transketol-ase of T. cruzi contains the PTS1 signal peptide, which possibly permits its distribution both in the glycosome and in the cytoplasm. The recombinant enzyme is apparently a dimer of 146 kDa (Igoillo-Esteve et al. 2007). Using the AnEnPi approach, we identified the sedoheptulose-bisphosphatase (EC: 3.1.3.37) that performs the addition of H20 to sedoheptulose 1,7-bisphosphate, resulting in sedoheptulose 7-phosphate and phosphate. This enzyme is a possible target for the development of drugs because they are specific to the parasite and absent in the human host. The Krebs cycle and oxidative phosphorylation - The intermediate energy metabolism that occurs in the mitochondria of T. cruzi was studied by several groups and reported several decades ago (von Brand & Agosin 1955, de Boiso & Stoppani 1973, Docampo et al. 1978, Rogerson & Gutteridge 1980). Cannata and Cazzulo (1984) reviewed the metabolism of carbohydrates and described, in detail, current knowledge of the Krebs cycle, as well as the process of aerobic fermentation in this parasite. Within this context, the main products of this peculiar metabolic pathway would be succinate from the reduction of fumarate coupled with the reduction of oxaloacetate and the generation of L-malate. More recently, the intermediary energy metabolism was reviewed by Urbina et al. (1993), including a discussion on the possibility of regulating the activity of the Krebs cycle according to the action of two glutamate dehydrogenases, one NAD+-dependent and the other NADP+ dependent, which indicates the use of amino acids as a main energy source for the parasite. The constitutive expression of the ATP-dependent phosphoenolpyruvate carboxykinase (PEPCK, EC 4.1.1.49) was described in all evolutionary forms of T. cruzi. The enzyme catalyses the reaction from phosphoenolpyruvate to oxaloacetate yielding ATP. In vertebrates, this enzyme is involved in glycogenesis, while in T. cruzi it appears to be involved in catabolism. Additionally, the inhibition of this enzyme by 3-mercaptopicolinic acid has been described in studies with the purified enzyme and in vivo (Urbina et al. 1990). This enzyme was also shown to have a specific requirement for transition metal ions that modulate the reactivity of a single essential thiol group, which differs from the hyper-reactive cysteines present in vertebrates or yeast (Jurado et al. 1996). Trapani et al. (2001) elucidated the 3D and dimeric structures of the PEPCK of T. cruzi, permitting progress in the study of this enzyme for the development of new therapeutic agents. Our data from in silico analysis indicate the presence of all the enzymes involved in the Krebs cycle, corroborating the analyses by KEGG, but also indicated a phosphoenolpyruvate carboxykinase (EC: 4.1.1.49) specific for T. cruzi and absent in humans (Table II, Fig. 2). Furthermore, the enzyme fumarate reductase (EC: 1.3.99.1) is also specific for T. cruzi and fumarate hydratase (EC: 4.2.1.2) in the parasite is analogous (Fig. 2) to the human protein. With relation to the mechanism of oxidative phosphorylation and the respiratory chain, our data and those of KEGG indicate the presence of all the multi-enzyme complexes in T. cruzi (Fig. 3), with several subunits that are either specific to the parasite or analogous to the human enzymes. Reports in the literature suggest that the process of oxidative phosphorylation in T. cruzi does not occur in a manner similar to humans, having been called oxidative fermentation (Cannata & Cazzulo 1984). Affranchino et al. (1985) conducted a study on the regulation of mitochondrial respiration in epimastigotes of T. cruzi, which showed, among other peculiarities, that the oxidation of NADH is not controlled by ADP. β-oxidation - The oxidation of fatty acids is an important source of ATP in many organisms, but this is apparently not the case for most parasites (van Hellemond & Tielens 2006). Initial analysis of the T. brucei genome (Berriman et al. 2005) but also of T. cruzi and Leishmania identified homologous genes for the four enzymes responsible for β-oxidation of fatty acids and this pathway probably occurs both in glycosomes and mitochondria (van Hellemond & Tielens 2006). The oxidation rates, however, seem minimal and oxidation can be for very specific fatty acids only or occur only under special conditions (Wiemer et al. 1996). The enzyme pyruvate phosphate dikinase was described in T. cruzi epimastigotes with a glycosomal localisation. The complete function of this enzyme is not completely understood but its reaction probably leads to pyruvate production from phosphoenolpyruvate and PPi (Acosta et al. 2004). The authors showed that palmitoyl-CoA β-oxidation occurs in glycosomes and suggest that this enzyme could be a link between glycolysis, fatty acid oxidation and the biosynthetic PPi-producing pathways in this organelle, as well as replacing pyrophosphatase in its classical thermodynamic role and eliminating the toxic PPi. Use of amino acids for energy production - T. cruzi epimastigotes metabolise asparagine, aspartate, glutamine, glutamate, leucine, isoleucine and proline (Sylvester & Krassner 1976). Alanine, aspartate and glutamate provide the Krebs cycle with intermediates (Silber et al. 2005), while the latter is an important intermediate in proline metabolism, which is an important carbon and energy source for the parasite. The amino group of glutamate can be transferred to pyruvate by both alanine aminotransferase and tyrosine aminotransferase, yielding a-ketoglutarate and alanine (Cazzulo 1994) or can be deaminated by glutamate dehydrogenase, which is localised in the cytoplasm and mitochondria (Duschak & Cazzulo 1991). A detailed computational analysis of the T. cruzi metabolic pathways involving amino acid metabolism was described previously and many instances of analogous enzymes in the parasite, compared to human, were pointed out (Guimarães et al. 2008). Many questions still remain open regarding energy metabolism in T. cruzi and our understanding of the physiological mechanisms involved in the generation of energy in this parasite. Furthermore, the identification of new therapeutic targets needs further work. The polyamine metabolism Due to the importance of polyamines for trypanosomatids, which are highly dependent on spermidine for growth and survival, polyamine metabolism has been well studied and several enzymes, such as arginase, ornithine decarboxylase (ODC), S-adenosylmethionine decarboxylase (AdoMetDC), spermidine synthase, trypanothione synthetase and trypanothione reductase have been targeted for the development of new drugs (Heby et al. 2007). T. cruzi does not contain ODC, an effective target of alpha-difluoromethylornithine for the treatment of sleeping sickness. Instead, the parasite salvages putrescine or spermidine from the host. Two candidate aminopropyltransferases have been proposed. Although T. cruzi maintains an apparently functional copy of the AdoMetDC, known inhibitors of this enzyme do not have much effect on the parasite (Beswick et al. 2006). The traditional cellular redox couple formed by the otherwise ubiquitous glutathione/glutathione reductase couple is replaced in trypanosomatids by the dithiol bis(glutathionyl)spermidine called trypanothione and the flavoenzyme trypanothione reductase. Trypanothione is the reducing agent of thioredoxin and tryparedoxin, which are small dithiol proteins that are reducing agents for the synthesis of deoxyribonucleotides, as well as for the detoxification of hydroperoxides by different peroxidases. The trypanothione reductase is an essential enzyme for the parasite and its absence in the mammalian host makes it an interesting target for drug development (Krauth-Siegel & Inhoff 2003, Martyn et al. 2007). More detailed analysis of biochemical pathways and increased high-throughput screening activities, using both synthetic compounds and natural products, set high hopes for the development of new drugs against Chagas disease, leishmaniasis and sleeping sickness. The many ongoing initiatives and the identification of numerous potential targets bring hope that a breakthrough in the treatment of these parasitic diseases will be shortcoming. REFERENCES
Copyright © 2009 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc09224t1.jpg] [oc09224f3.jpg] [oc09224t2.jpg] [oc09224f1.jpg] [oc09224f2.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}