 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
Memórias do Instituto Oswaldo Cruz, Vol. 104, No. 8, 2009, pp. 1194-1196 SHORT COMMUNICATIONS Further evaluation of an updated PCR assay for the detection of Schistosoma mansoni DNA in human stool samples Luciana I GomesI; Letícia HS MarquesI; Martin J EnkII; Paulo Marcos Z CoelhoII; Ana RabelloI, + ILaboratório
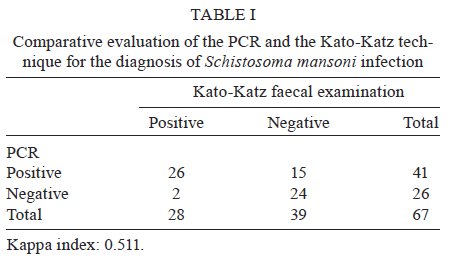
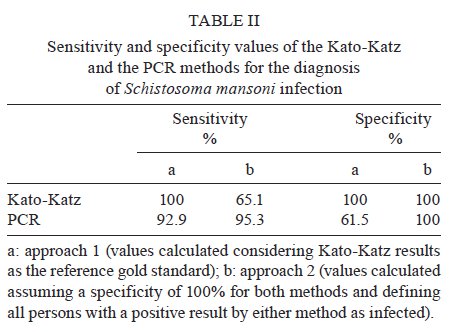
de Pesquisas Clínicas Financial support: FIOCRUZ (Program for Technical Development of Health Inputs), CNPq, FAPEMIG Received 12 August 2009 Code Number: oc09239 ABSTRACT A previously reported sensitive PCR assay for the detection of Schistosoma mansoni DNA was updated and evaluated. Changes in the DNA extraction method, including the use of a worldwide available commercial kit and the inclusion of additional quality control measures, increased the robustness of the test, as confirmed by the analysis of 67 faecal samples from an endemic area in Brazil. The PCR assay is at hand as a proven, reliable diagnostic test for the control of schistosomiasis in specific settings. Key words: schistosomiasis - molecular diagnosis - faecal samples - polymerase chain reaction Although the PCR technique has been extensively utilised for the diagnosis of numerous human diseases, its application for neglected diseases, especially schistosomiasis, has only recently been explored. Pontes et al. (2002) reported the first use of PCR for the diagnosis of Schistosoma mansoni DNA in faecal samples. High sensitivity and specificity rates were observed by this (Pontes et al. 2003) and another group (Allam et al. 2009) when compared to routine parasitological examination according to the Kato-Katz method (Katz et al. 1972). When using in-house techniques, the molecular methods may suffer from a lack of standardization and robustness. Both of these criteria are emphatically required for any possible application by control programs in the field, where decisions should not be made based on investigational results. In this note, the Schistosoma PCR assay is updated to explain the modifications introduced during the last few years that resulted in increased robustness and reproducibility. To further evaluate the modified method, 67 faecal specimens from a group composed of 27 children (female/male: 13/14; age range of 6-18 years) and 40 adult (female/male: 20/20; age range of 19-89 years) residents of Chonim de Cima, an endemic area in state of Minas Gerais, Brazil were analyzed. From the same sample, six slides were examined in search of parasite eggs by the parasitological Kato-Katz technique. This study was approved by the Ethical Committee of the Instituto de Pesquisas René Rachou-Fiocruz, Brazil (CEPSH: 03/2006). Individuals with positive faecal samples detected by the Kato-Katz technique were treated with a single oral dose of praziquantel (60 or 50 mg/kg for adults and children, respectively), according to the recommendation of the Brazilian Health Ministry. The alternative DNA extraction procedure used the QIAamp DNA Stool Mini Kit (Qiagen GmbH, Hilden, Germany) according to the manufacturer's recommendations and following the protocols DNA Isolation from Large Amounts of Stool and Isolation of DNA from Stool for Pathogen Detection. Total DNA was extracted from 500 mg of the 67 faecal specimens and from a saline solution containing ~2,000 eggs of S. mansoni, used for determining the detection limit of the PCR assay. The heating step was performed at 95ºC for 20 min. The concentration and purity of the DNA were determined spectrophotometrically by readings of A260 and A280 (Eppendorf, Hamburg, Germany). Faecal samples are known to be heterogeneous and therefore difficult to use in molecular analysis. In this study, the QIAamp® DNA Stool Mini Kit proved to be efficient. This worldwide available commercial kit allows the heating of the sample until 95ºC to facilitate the rupture of the egg and cellular lysis. It also includes Inhibitex, which adsorbs DNA-damaging substances and PCR inhibitors present in the faecal material. Thus, for amplification, the DNA samples were diluted only 5-fold with good reproducibility. DNA samples were amplified using the forward (5'-GATCTGAATCCGACCAACCG-3') and reverse (5'-ATATTAACGCCCACGCTCTC-3') primers designed by Pontes et al. (2002), which generate a 110 base pair product following the amplification procedure. For faecal amplification, DNA samples were diluted 5-fold and 2 μL were used as template. The same volume was used to amplify egg-derived DNA. The PCR was carried out in a final volume of 20 μL containing 2.0 U of Platinum Taq DNA Polymerase (Invitrogen, São Paulo, Brazil), 1X PCR Buffer (200 mM Tris-HCl, pH 8.4, 500 mM KCL - Invitrogen), 2.0 μg bovine serum albumin (BSA) (Sigma, Saint Louis, Missouri), 0.6 μM of (each) primer, 2.0 mM MgCl2 and 200 μM of each dNTP (Promega, Madison, WI). The PCR cycling program started with a pre-incubation at 95ºC for 5 min, followed by 14 cycles of 95ºC for 40 sec and 63ºC for 30 sec, 13 cycles of 80ºC for 40 sec and 63ºC for 30 sec, 11 cycles of 80ºC for 40 sec and 65ºC for 30 sec and a final elongation step at 72ºC for 5 min. The hot-start properties of the DNA polymerase used in this assay prevent the non-specific amplification that can occur due to polymerase activity at suboptimal temperatures during cycle initiation and guarantee a good analytical specificity. Furthermore, BSA was added to the PCR mix in order to stabilise the DNA polymerase and neutralise contaminating inhibitors. A two-stage cycling protocol was performed to improve the analytical sensitivity of the method. This protocol was chosen because most amplification occurs during the first stage (annealing at 63ºC) and a thermally separated second stage with higher annealing temperature (65ºC) for a lower number of cycles prevents high background due to the accumulation of primer oligomers. Drastic physical methods (separation of rooms and materials, use of bleach and laminar flux chamber with UV light) were applied during the procedure to minimise the chance of sample contamination. A carryover prevention method was also incorporated using uracil DNA glycosylase (UDG), an enzyme that cleaves the uracil base from the phosphodiester backbone of uracil-containing PCR products prior to amplification, without effect on natural (i.e., thymine-containing) DNA. Contaminating PCR products from previous laboratory experiments (in which dTTP was replaced with dUTP) are degraded by UDG, thus eliminating this potential contamination route (Longo et al. 1990, Schwab & McDevitt 2003). Consequently, 0.5 U of UDG (Invitrogen) and 400 μM dATP, dCTP and dGTP, 800 μM of dUTP (Promega) were added to the PCR master mix. The cycling program was preceded by a 10-min incubation at 37°C (UDG digestion period) and a 15-min incubation at 95ºC (Platinum Taq activation and UDG inactivation). Additionally, after the final cycle, the samples were held at 72°C or stored immediately at -20ºC to prevent residual UDG catalytic activity. As an internal quality control measure for the DNA isolation procedure and to test for the possible presence of PCR reaction inhibitors, all clinical samples were PCR-amplified for the human beta actin gene (ACTB) with the primers Aco1 and Aco2 (Musso et al. 1996). Five microlitres of the PCR-amplified material was subjected to electrophoresis on a 6% polyacrylamide gel and analyzed by silver staining. The PCR analytical sensitivity was evaluated using 10-fold serial dilutions ranging from 3 ng-300 ag of genomic DNA extracted from the eggs of S. mansoni. The detection limit was 3 fg of genomic DNA. Since the genome of S. mansoni contains ~580 fg of DNA, this is equivalent to less than a single cell of this multi-cellular parasite (Gomes et al. 2006). The efficiency of the UDG digestion system was determined through the addition of purified S. mansoni PCR product containing dUTP instead of dTTP (ranging from 0.2 ng-2 fg) to a new PCR reaction to mimic accidental contamination. The UDG system was able to prevent the subsequent reamplification of high levels (20 fg or 1.5 x 105 copies) of uracil-containing PCR product. This modified protocol did not change the performance of the method and maintained the detection limit of 3 fg of S. mansoni genomic DNA. Diagnostic parameters of the above-described new PCR assay procedure were established using faecal samples from 67 inhabitants of an endemic area in Brazil. The positivity ratio observed using the PCR technique (61.2%) was higher than that determined with the examination of six slides by the Kato-Katz technique (41.8%) (X2 = 5.05, p = 0.024). The geometric mean number of eggs per gram of faeces estimated by the Kato-Katz technique for the positive samples was 38 eggs per gram of faeces, which indicates a low intensity of infection. Table I depicts a direct comparison of the results obtained with the optimised PCR and the Kato-Katz technique. The Kappa index (Landis & Koch 1977) of 0.511 indicates a moderate agreement between the two methods. The analysis of discordant results revealed that 15 samples were positive only by the PCR and two samples were positive only by the Kato-Katz technique. These two patients had very low egg outputs (4 and 8 eggs per gram of faeces). A number of reasons may contribute to the two negative results of PCR, including variations in egg output and the uneven distribution of eggs in faeces (Engels et al. 1996). DNA degradation and the presence of PCR inhibitors, two other possible reasons, were eliminated, since both samples produced positive results for the amplification of the ACTB gene. Test parameters were calculated by two different approaches: (i) using results of the Kato-Katz method as reference for comparison or (ii) as an exercise, considering the specificity of both the Kato-Katz and PCR method as being 100% and assuming that an infected person is everyone with a positive result by either method (Table II). The sensitivity values of PCR were high irrespective of the reference considered: 92.9% (95% CI 75.0-98.8) for approach 1 and 95.3% (95% CI 82.9-99.2) for approach 2. The specificity values changed significantly depending on the reference used, with a value of 61.5% (95% CI 44.7-76.2) for approach 1. In approach 2, the Kato-Katz technique and PCR were considered to have specificities of 100%. The validity of this approach depends solely on the capability of the PCR to detect true positive samples, since that of the Kato-Katz technique has been established. However, all technical improvements described in this present study were carried out to increase the efficiency of PCR assay, contributing to a good analytical sensibility and specificity. Quality control measures, such as the use of DNA extraction and internal controls to prevent false-negative results, as well as the use of UDG to prevent carryover contamination and false-positive results, were adopted to ensure reliability to the assay. In conclusion, the updated Schistosoma PCR-assay can be considered as a quality assured diagnostic test for use by control programs in endemic areas where adequate infrastructure exists and the need for more sensitive diagnostic methods is critical. ACKNOWLEDGMENTS To Aureo Almeida and Ana Maria Pacheco, for the excellent technical assistance. REFERENCES
Copyright © 2009 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc09239t1.jpg] [oc09239t2.jpg] |
| |||||||||

{kind=link}
{kind=link}