 |
| About Bioline | All Journals | Testimonials | Membership | News |
|
||||||
|
||||||
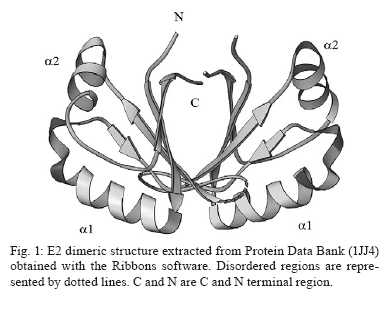
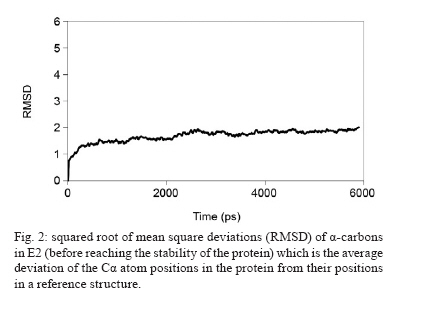
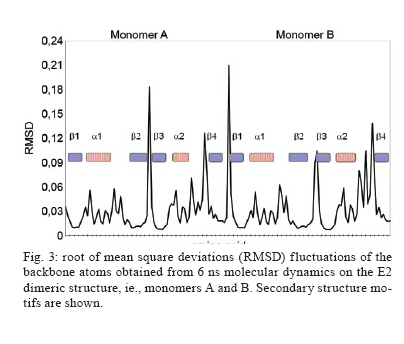
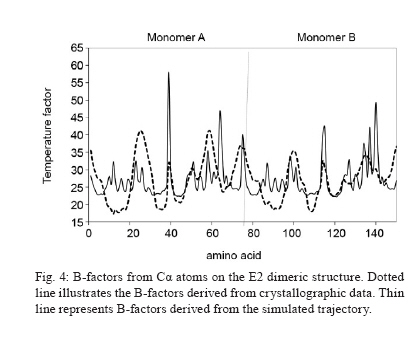
Memórias do Instituto Oswaldo Cruz, Vol. 105, No. 2, 2010, pp. 123-126 Assessing protein stability of the dimeric DNA-binding domain of E2 human papillomavirus 18 with molecular dynamics Raúl IseaI, +; José Luis RamírezII; Johan HoebekeIII ICentro de Biociencias Received 2 July 2009 Code Number: oc10021 ABSTRACT The objective of this study is to understand the structural flexibility and curvature of the E2 protein of human papillomavirus type 18 using molecular dynamics (6 ns). E2 is required for viral DNA replication and its disruption could be an anti-viral strategy. E2 is a dimer, with each monomer folding into a stable open-faced β-sandwich. We calculated the mobility of the E2 dimer and found that it was asymmetric. These different mobilities of E2 monomers suggest that drugs or vaccines could be targeted to the interface between the two monomers. Key words: human papillomavirus - molecular dynamics - DNA-binding domain - HPV-18 - protein stability Molecular epidemiology studies estimate that nearly 20% of the world's female population carries the human papillomavirus (HPV) in their cervixes, making this virus the most frequently sexually transmitted infection (Jansen & Shaw 2004). The correlation between HPV infections and uterine cancer has been firmly established (Schiller & Lowy 2006) and in some countries this disease is the leading cause of death in the female population (Cavalcanti et al. 1996, Rivera et al. 2002, Lepique et al. 2009). HPV belongs to the Papovaviridae family and its genome is a circular, double-stranded DNA molecule of 8 kbp in length. During the early phase of infection, HPV expresses a tandem of genes from E1-E8, which are involved in the regulation of viral replication; in the late phase of infection, two additional genes, L1 and L2, which are responsible for viral capsid assembly, are expressed (Okun et al. 2001). E2 also acts as a transcriptional repressor of E6 and E7 when it binds the early promoter (Desaintes et al. 1999). As E2 plays a key role in HPV replication, this protein has been suggested as a potential target for vaccine development (Schiller & Hidesheim 2000). This suggestion is supported by the fact that vaccination of rabbits with a DNA fragment encoding cotton-tail rabbit papillomavirus E2 protein protects inoculated animals against subsequent tumour challenges (Govan et al. 2006). For this reason, we decided to examine the tertiary structural stability of E2 in HPV-18, a virus implicated in cervix carcinoma, aiming to identify the zones that could be more susceptible to drug interactions or vaccine production. This could help in the development of a screening assay for new antiviral drugs. In addition, E2 plays a key role in inducing apoptosis by a p53-independent mechanism (Desaintes et al. 1999). Using X-ray diffraction, the tertiary structure of the DNA-binding domain of HPV-18 E2 was determined both as an E2-DNA complex (PDB: 1JJ4) and as an E2 domain without DNA (PDB: 1F9F). In both cases, the E2 quaternary structure was a dimer (Fig. 1) and each monomer was made up of four antiparallel β-strands and two α-helices. The monomer topology was typically described as β1-α1-β2-β3-α2-β4 (Fig. 1), whereas the total structure of the E2 protein DNA-binding domain was of a β-barrel type. As explained previously, the α1 helix is involved in the interaction with the viral DNA and could be the recognition helix (Hegde 2002). MATERIALS AND METHODS To start molecular dynamics (MD) simulations, we obtained from the Protein Data Bank (PDB) (http://www.pdb.org) the E2 DNA-binding domain coordinates either as a complex with DNA (PDB code: 1JJ4) or in its unbound conformation (PDB code: 1F9F). In the case of the complexed E2-DNA region (1JJ4), we only used the protein coordinates by subtracting the coordinates of the DNA molecule. We thus had two starting points to study the mobility of the E2 protein. In the calculations, the amino acids 283-284 and 323-327 were not included (Kim et al. 2000) because they were not allocated in the results of the X-ray diffraction studies and their inclusion could thus lead to potentially erroneous calculations. We shall, however, discuss the potential impact of this omission on our conclusions. MD simulations were done with the MD program NAMD version 2.5 (Kale et al. 1999), using a cluster under the Linux operating system, with a 1-fs time step. The total time of the molecular dynamic simulations was 6 ns. The force field used was CHARMM version 27 (Mackerell et al. 1998). Non-covalent and van der Waals energies were calculated with a cut-off distance of 12 Å. Solvation was simulated with a water layer of at least 5 Å around the protein, in a 45 x 48 x 110-Å box, containing 2,583 water molecules under periodic boundary conditions. To neutralise the charge of the protein, 10 chloride (Cl-) counter-ions were inserted (the total number of atoms was 10,333). The protonation states of the ionisable residues were assigned and used in the ShakeH algorithm implemented in the VMD software to fix the bond between each hydrogen and its mother atom to the nominal bond length (Humphrey et al. 1996). Explicit velocity and position Verlet-like algorithms of the 2nd order were proposed to integrate the equations of motion (Ryckaert et al. 1997). We used a method of energy minimisation, using the steepest descent algorithm (preceded by a position-restrained stage for protein atoms) and a conjugate gradient, until an energy gradient less than 1.0 kcal/mol/Å was reached. The MD simulations were performed according to the following criteria: 50 ps with the positions of the protein's atoms restrained to permit solvent equilibration, 150 ps with the positions of the backbone's protein atoms restrained to permit the gradual liberation of the system and then a full MD for 6 ns without restriction (NPT ensemble was completed). The B-factors derived from the trajectory were calculated as 8/3π2 <|Δr|2>, where <|Δr|2> is the mean square atomic displacement relative to the average position from all trajectories of MD. RESULTS An initial examination of the input data revealed that both structures were very similar, that is, the distance between the Glu340 in both monomers was 24.8 Å in 1JJ4 (after subtraction of the DNA coordinates), while in the E2 protein without DNA, the distance was up to 27.3 Å (an increase of < 10%). The angle formed by both helices (Lys308-Leu347-Glu340) was 106.3º for 1JJ4, while the angle decreased less than 8% to 97.9º for the Arg307-Leu347-Glu340 in the domain without DNA. Therefore, we can conclude that the DNA-interacting domain does not change significantly during the formation of the complex. If this is the case, we can monitor these parameters and determine whether they are in the range of correct values. Taking this approach and using MD simulations, we calculated the HPV-18 E2 protein mobility. The root mean square deviation (RMSD) values of all atoms were calculated from the initial X-ray coordinates until 6 ns simulation was completed (Fig. 1). A close inspection of the trajectory shows that the highest RMSD values detected were 1.94 Å (for PDB 1JJ4) and 1.78 Å (for 1F9F). The RMSD average values were 1.76 Å and 1.42 Å obtained from MD up to 6 ns when using the initial formations derived from 1JJ4 (Fig. 2) before reaching the stability of the protein) and 1F9F, respectively (data not shown), where the median quadratic deviation among amino acids was used to calculate the mobility of Cα atoms. A different study with calculations performed for 6 ns could be done, but the range used here was sufficient to determine the dimer's stability. This is because the link angle formed by Glu340 with both monomers oscillated around 24.6 Å (22.32 for one limit and 27.43 for the other) a range contained in the DNA-free 1F9F formation values (from 21.12-28.21 Å). In addition, the angle formed by Lys308-Leu347-Glu340 along the dynamic was 108.5°, with a minimum value of 97.0 and an upper limit of 123.6 (the values for the ligand-free protein ranged from 101.2-129.2°). Although these results suggest that the missing amino acids from the X-ray determination (i.e., 283, 284 and 323-327) do not contribute to dimer stability, this conclusion must be verified by further studies based on free-energy calculations. At the same time, the mobility associated with the amino acids forming the α1 region (from 297-308 in the 1JJ4 and 296-307 in the 1F9F protein) of both monomers had the same behaviour, whereas the mobility of the connecting regions between secondary structures β2-β3 and α2-β4 were lower than for monomer B. We observed a low mobility of amino acids forming the β-sheet structures that lacked the small oscillation observed for the α-helix structures (Fig. 3). In general, the E2 protein β-barrels were very stable. Fig. 4 shows a comparison between the B-factors derived from the simulation (for Cα atoms only) and from crystallographic studies. Most of the mean square displacement was concentrated in the region between β2-β 3 and α2-β 4. The crystallographic B-factors did not display any meaningful increase at this particular region. Therefore, we concluded that in the HPV-18 E2 protein, relatively large molecular motions are mostly restricted to the A monomer and this observation agrees with the values of B-factor temperature obtained from X-rays. Analysis of RMSD showed that the helices in this region were highly stable and, consequently, we believe that the large RMSD values were not due to the unfolding of these structures. The region α1-β2 had low mobility, suggesting high stability during the dynamics. Finally, previous work (Hajduk et al. 1997) suggests that a possible strategy to inhibit the interaction of E2 dimers with DNA is precisely by blocking residues Leu306, Leu309, Leu313, Val358, Ile360 and Pro361. Therefore, we calculated the RMSD of those amino acids and obtained the same behaviour as for residues 297-301, the values of RMSD for each residue being 0.02, 0.02, 0.01, 0.05, 0.03 and 0.02, respectively. DISCUSSION In this paper, we analysed the dynamic behaviour of the HPV-18 E2 protein in two different formation schemes, which corresponded to the E2 coordinates when it was crystallised with or without DNA. Both calculations were performed by the same MD model, showing that the mobility was asymmetric through RMSD calculations (Fig. 3). Although we cannot exclude the possibility that the deleted residues could affect the mobility, our conclusions regarding the asymmetric mobility remain valid because the deleted loop is present in both monomers. Zimmerman and Maher (2003) proposed that the E2-binding affinity of a DNA site depends on the tetranucleotide stretch (NNNN) in the sequence 5'-ACCGNNNNCGGT-3', responsible for the DNA curvature in the minor groove. Moreover, no base-specific contacts could be detected in the crystal structure. Both pieces of evidence suggest flexibility in the protein binding site, confirming that the calculated mobility is functionally important. In addition, the RMSD calculated from MD simulations agreed with the B-factors obtained by X-ray diffraction, the mobility being asymmetric between monomers (Fig. 4). The lack of good electron density maps due to the high B-factor in the unbound state strengthens the hypothesis that the disordered state is required to allow E2 to interact with DNA to form a more stable protein-DNA complex. Kim et al. (2000) indicated that in this region the tertiary structure of the E2 DNA-binding site is disordered and this change in the helix curvature may therefore help to lower the interaction energy of the E2-DNA complex. We can also speculate that the asymmetric mobility favours the contact of E2 with the DNA minor and major grooves and that this interaction is very stable because no protein unfolding was detected during the 6 ns of MD. This, however, does not exclude the possibility that the protein-DNA complex could lead to dimer dissociation, as suggested by Lima et al. (2000). ACKNOWLEDGEMENTS To Sharon Sumpter, for all comments about this paper, and to the editor and referees, for their essential contributions of evaluating this paper. This paper is dedicated to the memory of Leonardo Mateu Suay who died on 29th June 2008. REFERENCES
Copyright © 2010 - Instituto Oswaldo Cruz - Fiocruz The following images related to this document are available:Photo images[oc10021f2.jpg] [oc10021f1.jpg] [oc10021f3.jpg] [oc10021f4.jpg] |
| |||||||||

{kind=link}
{kind=link}
{kind=link}
{kind=link}